José Luis Oliveira
Universidade de Aveiro, DETI / IEETA
3810-193 Aveiro, Portugal
jlo@ua.pt
(+351) 234 370 500
GTO
 About
AboutGTO is a toolkit for genomics and proteomics, namely for FASTQ, FASTA and SEQ formats, with many complementary tools. The toolkit is for Unix-based systems, built for ultra-fast computations. GTO supports pipes for easy integration with the sub-programs belonging to GTO as well as external tools. GTO works as LEGOs, since it allows the construction of multiple pipelines with many combinations. GTO includes tools for information display, randomisation, edition, conversion, extraction, search, calculation, compression, simulation and visualisation. GTO is prepared to deal with very large datasets, typically in the scale of Gigabytes or Terabytes (but not limited).
Citation DownloadNETDIAMOND Platform
Masters thesis (Tiago Almeida)
Tiago Almeida, “Neural Information Retrieval for Biomedical Question-Answering”
July 2019
PhD Defense (Eduardo Pinho)
Eduardo Miguel Coutinho Gomes de Pinho, “Multimodal Information Retrieval in Medical Imaging Repositories”
July 2019
D4 – Deep Drug Discovery and Deployment
Funding entity: FCT Period: 2018-2021
Funding entity: FCT Period: 2018-2021 D4 proposes the use of state-of-the-art Deep Learning methods to tackle the challenges identified in each of the initial stages of the drug discovery pipeline. The main contribution of this project is the creation of an improved computational pipeline that uses Deep Learning architectures to support the drug discovery process. The pipeline will be implemented within a framework that will be available to the community. Both the final platform and the computational methods will be validated with the close collaboration of the industrial partner, which will apply it to develop novel therapeutics for neurodegenerative amyloid diseases.EHDEN – European Health Data & Evidence Network
Funding entity: H2020/IMI-JU Period: 2018-2023
Funding entity: H2020/IMI-JU Period: 2018-2023 Presently, Europe is generating unprecedented amounts of patient-level information contained in Electronic Health Record (EHR) systems and other types of health databases. This includes structured data in the form of diagnoses, medication, laboratory test results, etc., and unstructured data in clinical narratives, all of which likely contain invaluable insights into the natural history and burden of disease, its clinical management and outcomes, and wider perspectives on both healthcare and the patient experience of it. It is our ambition to fully leverage these vast volumes of data to improve clinical practice and individual patient outcomes by increasing our understanding of disease and treatment pathways and effects. We will galvanize transparent and reproducible analytics that will generate valid real-world evidence to improve patient care and enable medical outcomes-based research at an unprecedented scale. The Electronic Health Data and Evidence Network (EHDEN) consortium will provide the infrastructure and eco-system to make this ambition come true, supporting the disease-specific projects in the IMI Big Data for Better Outcomes (BD4BO) programme, academia, pharmaceutical, and life sciences, regulatory and allied institutions.PhD Defense (Fernanda Correia)
Fernanda Brito Correia, “Prediction And Analysis Of Biological Networks Structure And Dynamics”
April 2019
Masters thesis defense (João Almeida)
João Rafael Almeida, “Software solution for clinical protocol management”
18 Jul, 18.00h
18th Portugaliæ Genetica 2018 – Best Poster Award
The work entitled “eQTL analysis of the NAPRT locus” won the Best Poster Award at the 18th Portugaliæ Genetica – Genetic Diversity in Structure and Regulation, that was held on 22-23rd of March 2018 in Porto. The work was carried out by PhD. student Sara Duarte-Pereira, researcher Sérgio Matos, Professors José Luís Oliveira and Raquel M. Silva from the Institute of Electronics and Informatics Engineering of Aveiro (IEETA).
18th Portugaliæ Genetica 2018 – Best Oral Presentation Award
Diogo Pratas was granted Best Oral Presentation Award at the 18th Portugaliæ Genetica – Genetic Diversity in Structure and Regulation, held on 22-23rd of March 2018 in Porto. The work presented is entitled “Metagenomic composition analysis of ancient DNA samples”.
DICOM Validator
The DICOM validator is a web-based solution for evaluation the compliance of PACS applications with the DICOM standard. It features the “as-a-service” business model, which allows users to immediately reach their goals without the extensive setup efforts required by similar solutions. The DICOM Validator is also a community-driven initiative, where users all around the world are invited to contribute to the creation and maintenance of the DICOM module definitions. With your help, we will soon reach full coverage of the DICOM Standard and keep-up with its latest revisions.
![]()
Check us at: https://bioinformatics.ua.pt/dicomvalidator/
SOCA – Smart Open Campus
Funding entity: Centro 2020
Period: 2017-2020
The sensing of the person in physical context enables personalized and predictive responses, and is a major step towards a smarter and safer environment. The main objective of SOCA is to create an open innovation ecosystem where data is gathered from multiple sources, processed, integrated, and made available for applications and users, and that is able to create a service sphere able to assist every individual inside it – from personal health to routine daily chores. For this endeavor, the academic campus will provide the perfect framework to support and trial innovations on the smart city and on the assisted living arenas.
SCALEUS
 SCALEUS is a data migration tool that can be used on top of traditional systems to enable semantic web features. This user-friendly tool help users easily create new semantic web applications from scratch. Targeted at the biomedical domain, this web-based platform offers, in a single package, a high-perfomance database, data integration algorithms and optimized text searches over the indexed resources. SCALEUS is available as open source at http://bioinformatics-ua.github.io/scaleus/.
SCALEUS is a data migration tool that can be used on top of traditional systems to enable semantic web features. This user-friendly tool help users easily create new semantic web applications from scratch. Targeted at the biomedical domain, this web-based platform offers, in a single package, a high-perfomance database, data integration algorithms and optimized text searches over the indexed resources. SCALEUS is available as open source at http://bioinformatics-ua.github.io/scaleus/.
Ann2RDF
 Ann2RDF is an interoperable semantic layer that unifies text-mining results originated from different tools, information extracted by curators, and baseline data already available in reference knowledge bases, enabling a proper exploration using semantic web technologies. This result in a more suitable transition process, in which desired annotations are enriched with the possibility to be shared, compared and reused across semantic Knowledge Bases. Ann2RDF is available at http://bioinformatics-ua.github.io/ann2rdf/.
Ann2RDF is an interoperable semantic layer that unifies text-mining results originated from different tools, information extracted by curators, and baseline data already available in reference knowledge bases, enabling a proper exploration using semantic web technologies. This result in a more suitable transition process, in which desired annotations are enriched with the possibility to be shared, compared and reused across semantic Knowledge Bases. Ann2RDF is available at http://bioinformatics-ua.github.io/ann2rdf/.
I2X

I2X is a reactive and event-driven framework that simplifies and automates real-time data integration and interoperability. This platform streamlines the creation of customizable integration tasks connecting heterogeneous data sources with any kind of services. Integration is poll-based, with intelligent agents monitoring data sources, or push-based, where the platform waits for data submission by external resources. I2X delivers data to services through a comprehensive template engine, where the platform maps data from the original data source to the destination resources. I2X is an open-source framework available online at https://bioinformatics.ua.pt/i2x/.
TASKA
Task management systems are crucial tools in modern organizations, by simplifying the coordination of teams and their work. Those tools were developed mainly for task scheduling, assignment, follow-up, and accountability. On the other hand, scientific workflow systems also appeared to help putting together a set of computational processes through the pipeline of inputs and outputs from each, creating in the end a more complex processing workflow. However, there is sometimes a lack of solutions that combine both manually operated tasks with automatic processes, in the same workflow system.
TASKA is a web-based platform that incorporates some of the best functionalities of both systems, addressing the collaborative needs of a task manager with well-structured computational pipelines.
The system is currently being used by EMIF (European Medical Information Framework) for the coordination of clinical studies.
A demo installation of TASKA is available online at https://bioinformatics.ua.pt/taska
MONTRA
![]()
MONTRA is a rapid-application development framework designed to facilitate the integration and discovery of heterogeneous objects which may be characterized by distinct data structures. Initially designed as a framework which allows biomedical researchers to easily set up dynamic workspaces, where they can publish and share sensitive information about their data entities, MONTRA is suitable for any data domain, by allowing the characterisation of the most diverse entities or group of entities (datasets). Through the use of a common skeleton, it automatically generates a fully-fledged web data catalogue, ensuring data privacy protection.
MONTRA is being used by several European projects, and its source code is publicly available at https://github.com/bioinformatics-ua/montra.
NETDIAMOND – New Targets in Diastolic Heart Failure: from Comorbidities to Personalized Medicine
Funding entity:P2020/PAC
Period: 2016-2019
Heart failure (HF) is a highly prevalent syndrome of impaired cardiac function that constitutes the main cause of hospitalization and disability amongst the elderly, a leading cause of mortality, morbidity and resource consumption. HF with preserved ejection fraction (HFpEF) is characterized by preserved ejection, impaired cardiac filling, lung congestion and effort intolerance, accounting for a rising proportion of over 50% of cases due to ageing and increasing incidences of systemic arterial hypertension (SAH), obesity and diabetes mellitus (DM). The current proposal sets forth to address this issue by a mixed strategy of discovery science approach through comprehensive multi-omics studies in plasma and tissues from HFpEF patients and animal models with and without comorbidities (DM, SAH and obesity), and an hypothesis-driven approach focusing on disturbances of cell function and communication in endothelial cells (EC), cardiac fibroblasts (CF), adipocytes and CM. A holistic view of HFpEF and of the role of comorbidities will be achieved by correlating and integrating transcriptomics, proteomics and lipidomics studies with clinical data. The impact on CM and myocardium will be comprehensively assessed in vitro and in vivo. Finally, preclinical testing of functional foods, synthetic antioxidants, enhanced bioavailability putative therapeutic molecules as well as other potentially effective gene targets identified along the project’s course will be assayed.
Multimodal Information Retrieval in Medical Imaging Repositories
Funding entity:FCT
Period: 2016-2019
Digital medical imaging systems are, nowadays, essential tools in clinical practice, both in decision supporting and in treatment management. The main objective of this project is to investigate new solutions for extracting, merging and searching over multimodal data, including text (DICOM metadata and diagnosis reports) and image information. Relevance feedback will be also investigated to increase the results quality of the proposed multimodal architecture. It is also our aim to investigate the contribution of semantic information in imaging retrieval and information extraction. We will develop a semantic PACS concept to provide search functionality using context-dependent semantic information.
SCREEN-DR – Image Analysis and Machine Learning Platform for Innovation in Diabetic Retinopathy Screening
Funding entity:FCT (CMU-Portugal)
Period: 2016-2019
Diabetic Retinopathy (DR) is a leading cause of blindness in the industrialized world that can be avoided with early treatment, demanding an earlier diagnosis in a stage where the treatment is still possible and effective. DR evolves silently without any visual symptoms, during the early stages of the disease.
Under this context, the vision of the consortium SCREEN-DR is to create a distributed and automatic screening platform for DR, based on the state-of-the-art Information and Communication Technologies (ICT), including advanced Picture Archiving and Communication Systems (PACS) management, Machine Learning and Image Analysis, enabling immediate response from health carers, allowing accurate follow-up strategies, and fostering technological innovation.
rebico
 About
AboutA survey on data compression methods for:
- protein sequences
- genomic sequences:
- reference-free
- reference-based
- specific formats:
- FASTA
- FASTQ
- SAM/BAM
CitationM. Hosseini, D. Pratas, A. J. Pinho. “A survey on data compression methods for biological sequences.” Information 7.4 (2016): 56.
DownloadMasters thesis defense (Ricardo Ribeiro)
Ricardo Filipe Gonçalves Ribeiro, “TASKA: A modular and easily extendable system for repeatable workflows”
23 Mai, 14.30 pm
GeCo
AboutGeCo is a method and tool designed for the compression and analysis of genomic data. As a compression tool, GeCo is able to provide additional compression gains over several top specific tools in different levels of redundancy. As an analysis tool, GeCo is able to determine absolute measures, namely for many distance computations, and local measures, such as the information content contained in each element, providing a way to quantify and locate specific genomic events. GeCo can afford individual compression and referential compression (conditional or conditional exclusive). The tool is memory adjustable, using hash-caches for the deepest context models, making possible to be run in modest computers.
CitationD. Pratas, A. J. Pinho, P. J. S. G. Ferreira. Efficient compression of genomic sequences. Proc. of the Data Compression Conference, DCC-2016, Snowbird, UT, March 2016. (accepted)
DownloadPhD Defense (Luis Bastião)
Luis Bastião Silva, “A federated architecture for biomedical data integration”
Universidade de Aveiro, DETI/IEETA
smash
 About
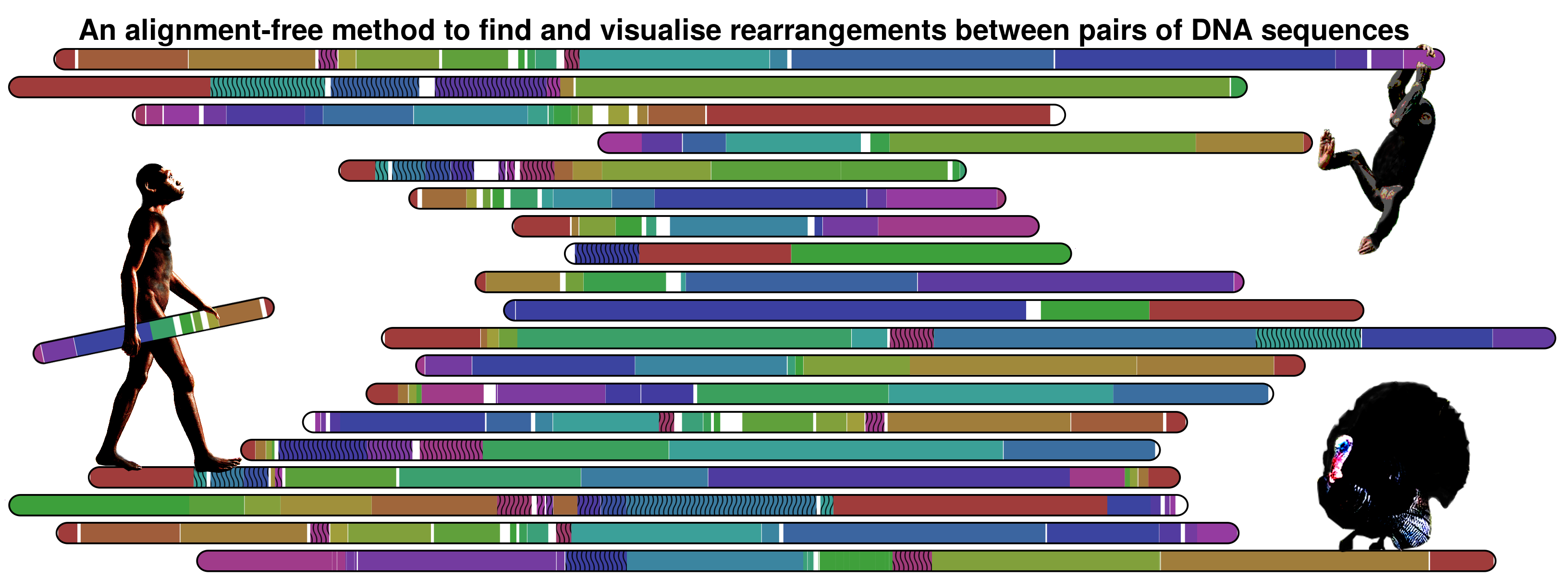
AboutSmash is a completely alignment-free method/tool to find and visualise genomic rearrangements. The detection is based on conditional exclusive compression, namely using a FCM (Markov model), of high context order (typically 20). For visualisation, Smash outputs a SVG image, with an ideogram output architecture, where the patterns are represented with several HSV values (only value varies). The method can perform both in small- and large-scale. Nevertheless is more directed to large-scale since that the main aim of the research is to know where the large-scale [chromosomal by chromosome] of several primates was equal/different, having at a glance a map of the entire genomes. Therefore the method aims to solve evolutionary species Rubik’s cube. The following image, illustrating the information maps between human and chimpanzee for the several chromosomes, depicts such an example study:


Nevertheless, the method is not limited to primates information. The following image show the information map between Meleagris gallopavo and Gallus gallus chromosomes 1 using a threshold of 0.95.
Download CitationDiogo Pratas, Raquel M. Silva, Armando J. Pinho, Paulo J. S. G. Ferreira. An alignment-free method to find and visualise rearrangements between pairs of DNA sequences. Sci. Rep. 5, 10203 (2015); doi:10.1038/srep10203.
PhD Defense (Carlos Ferreira)
Carlos Ferreira, “Handling Data Access Latency in Distributed Medical Imaging Environments”
Universidade de Aveiro, DETI/IEETA
Date: 2015.04.10, 10.00 AM
Anfiteatro, Reitoria, Universiade de Aveiro
eagle
 EAGLE: alignment-free method to compute relative absent words (RAWs)
EAGLE: alignment-free method to compute relative absent words (RAWs)
AboutEAGLE is an alignment-free method and associated program to compute relative absent words (RAW) in genomic sequences using a reference sequence. Currently, EAGLE runs on a command line linux environment, building an image with patterns reporting the absent words regions (in SVG) as well as reporting the associated positions into a file. EAGLE has got scripts to run on the current outbreak and the other existing ebola virus genomes (using the human as a reference), including the download, filtering and processing of the entire data.
CitationRaquel M. Silva, Diogo Pratas, Luísa Castro, Armando J. Pinho & Paulo J. S. G. Ferreira. Bioinformatics (2015): btv189.
DOI: 10.1093/bioinformatics/btv189.
DownloadPhD Defense (Paulo Gaspar)
Paulo Gaspar, “Computational methods for gene characterization and genome knowledge extraction”
Universidade de Aveiro, DETI/IEETA
MENT
 MENT: Microarray comprEssiOn Tools
MENT: Microarray comprEssiOn Tools
AboutMENT is a set of tools for lossless compression of microarray images, however, it can be used in other kind of images such as medical, RNAi, etc. This set of tools is divided into two categories, defined by the decomposition approach used:
- Bitplane Decomposition
- Binary-Tree Decomposition
In what follows, we will describe the set of tools available in MENT:
- BOSC06 (Bitplane decOmpoSition Compressor 2006) – Lossless compression tool for microarray images introduced by Neves and Pinho in 2006 (Neves 2006). This tool uses an image-INDEPENDENT context configuration and arithmetic coding.
- BOSC09 (Bitplane decOmpoSition Compressor 2009) – Lossless compression tool for microarray images introduced by Neves and Pinho in 2009 (Neves 2009). This tool uses an image-DEPENDENT context configuration and arithmetic coding.
- BOSC09HC (Bitplane decOmpoSition Compressor 2009 using Histogram Compaction) – Tool inspired on method introduced by (Neves 2009) where it was added an Histogram Compaction unit in order to remove some redundant bitplanes. This Histogram Compaction is usefull for images that have a reduced number of intensities.
- BOSC09SBR (Bitplane decOmpoSition Compressor 2009 using Scalable Bitplane Reduction) – Tool inspired on method introduced by (Neves 2009) where it was added an Scalable Bitplane Reduction unit in order to remove some redundant bitplanes. The Scalable Bitplane Reduction technique was first introduced by Yoo 1999.
- SBC (Simple Bitplane Coding) – Tool inspired on one of Kikuchi’s work (Kikuchi 2009, Kikuchi 2012).
- BOSC09MixSBC (Bitplane decOmpoSition Compressor 2009 Mixture with Simple Bitplane Coding) – Tool based on a mixture of finite-context models. In this particular case, we only considered two different models. The first one used by Neves and Pinho (Neves 2009) and the other one based on a Simple Bitplane Coding inspired on Kikuchi’s work (Kikuchi 2009, Kikuchi 2012).
- BITTOC (Binary Tree decomposiTiOn Compressor) – Tool inspired on Chen’s work regarding compression of color-quantized images (Chen 2002). This tool performance was studied in the context of medical images by Pinho and Neves in 2009 (Pinho 2009) and more recently applied to microarray images (Matos 2014).
- CmpImgs (Compare Images) – An image comparasion tool.
Microarray image sets- ApoA1 (32 images | 66.4MB | download)
- Arizona (6 images | 694.9MB | download)
- IBB (44 images | 1.03GB | download)
- ISREC (14 images | 26.7MB | download)
- Omnibus – Low Mode (25 images | 2.5GB | download)
- Omnibus – High Mode (25 images | 2.5GB | download)
- Stanford (40 images | 396MB | download)
- Yeast (109 images | 219MB | download)
- YuLou (3 images | 40.7MB | download)
CitationIf you use some tool from MENT, please cite the following publications:
- Luís M. O. Matos, António J. R. Neves, Armando J. Pinho, “Lossy-to-lossless compression of biomedical images based on image decomposition”,in Applications of Digital Signal Processing through Practical Approach, Sudhakar Radhakrishnan (Editor), InTech, pp. 125-158, October 2015. DOI: doi.org/10.5772/60650
- Luís M. O. Matos, António J. R. Neves, Armando J. Pinho, “A rate-distortion study on microarray image compression”, in Proceedings of the 20th Portuguese Conference on Pattern Recognition, RecPad 2014, Covilhã, Portugal, October 2014. DOI: doi.org/10.13140/2.1.3431.2969
- Luís M. O. Matos, António J. R. Neves, Armando J. Pinho, “Compression of microarrays images using a binary tree decomposition”, in Proceedings of the 22nd European Signal Processing Conference, EUSIPCO 2014, Lisbon, Portugal, September 2014. DOI: doi.org/10.13140/2.1.1980.5761
- Luís M. O. Matos, António J. R. Neves, Armando J. Pinho, “Compression of DNA microarrays using a mixture of finite-context models”, in Proceedings of the 18th Portuguese Conference on Pattern Recognition, RecPad 2012, Coimbra, Portugal, October 2012. DOI: doi.org/10.13140/2.1.1061.8245
- Luís M. O. Matos, António J. R. Neves, Armando J. Pinho, “Lossy-to-lossless compression of microarrays images using expectation pixel values”, in Proceedings of the 17th Portuguese Conference on Pattern Recognition, RecPad 2011, Porto, Portugal, October 2011. DOI: doi.org/10.13140/2.1.3553.4403
- Luís M. O. Matos, António J. R. Neves, Armando J. Pinho, “Lossless compression of microarrays images based on background/foreground separation”, in Proceedings of the 16th Portuguese Conference on Pattern Recognition, RecPad 2010, Vila Real, Portugal, October 2010. DOI: doi.org/10.13140/2.1.3815.5843
- António J. R. Neves, Armando J. Pinho, “Lossless compression of microarray images using image-dependent finite-context models”, in IEEE Transactions on Medical Imaging, volume 28, number 2, pages 194-201, February 2009. DOI: dx.doi.org/10.1109/TMI.2008.929095
- António J. R. Neves, Armando J. Pinho, “Lossless Compression of Microarray Images”, in Proceedings of the IEEE International Conference on Image Processing, ICIP-2006, Atlanta, GA, pages 2505-2508, 8-11 October, 2006. DOI: dx.doi.org/10.1109/ICIP.2006.31280
DownloadXS
 XS: a FASTQ read simulator
XS: a FASTQ read simulator
AboutXS is a skilled FASTQ read simulation tool, flexible, portable (does not need a reference sequence) and tunable in terms of sequence complexity. XS handles Ion Torrent, Roche-454, Illumina and ABI-SOLiD simulation sequencing types. It has several running modes, depending on the time and memory available, and is aimed at testing computing infrastructures, namely cloud computing of large-scale projects, and testing FASTQ compression algorithms. Moreover, XS offers the possibility of simulating the three main FASTQ components individually (headers, DNA sequences and quality-scores). Quality-scores can be simulated using uniform and Gaussian distributions.
CitationPratas, D., Pinho, A. J., & Rodrigues, J. M. R. (2014). XS: a FASTQ read simulator. BMC research notes, 7(1), 40.
Web download Download and install from consolewget https://github.com/pratas/xs/archive/master.zip
unzip master.zip
cd xs-master
make
unzip master.zip
cd xs-master
make
Or alternatively:
wget https://bioinformatics.ua.pt/wp-content/uploads/2014/02/XS.tar.gz
tar -vzxf XS.tar.gz
cd XS
make
SACO
 SACO: a lossless compression tool for the sequences alignments found in the MAF files.
SACO: a lossless compression tool for the sequences alignments found in the MAF files.
AboutSACO was designed to handle the DNA bases and gap symbols that can be found in MAF files. Our method is based on a mixture of finite-context models. Contrarily a recent approach, it addresses both the DNA bases and gap symbols at once, better exploring the existing correlations. For comparison with previous methods, our algorithm was tested in the multiz28way dataset. On average, it attained 0.94 bits per symbol, approximately 7% better than the previous best, for a similar computational complexity. We also tested the model in the most recent dataset, multiz46way. In this dataset, that contains alignments of 46 different species, our compression model achieved an average of 0.72 bits per MSA block symbol.
Data sets CitationIf you use this software, please cite the following publications:
- Luís M. O. Matos, Diogo Pratas, and Armando J. Pinho, “A Compression Model for DNA Multiple Sequence Alignment Blocks”, in IEEE Transactions on Information Theory, volume 59, number 5, pages 3189-3198, May 2013. DOI: dx.doi.org/10.1109/TIT.2012.2236605
- Luís M. O. Matos, Diogo Pratas, and Armando J. Pinho, “Compression of whole genome alignments using a mixture of finite-context models”, in Proceedings of the International Conference on Image Analysis and Recognition, ICIAR 2012, (Editors: A. Campilho and M. Kamel, volume 2324 of Lecture Notes in Computer Science (LNCS)), pages 359-366, Springer Berlin Heidelberg, Aveiro, Portugal, June 2012. DOI: doi.org/10.1007/978-3-642-31295-3_42
DownloadMAFCO
 MAFCO: a compression tool for MAF files
MAFCO: a compression tool for MAF files
AboutMAFCO is a lossless compression tool specifically designed to compress MAF (Multiple Alignment Format) files. Compared to gzip, the proposed tool attains a compression gain from ≈ 34% to ≈ 57%, depending on the data set. When compared to a recent dedicated method, which is not compatible with some data sets, the compression gain of MAFCO is about 9%. MAFCO was designed and implemented at IEETA, a research unit of the University of Aveiro, and is available for non-commercial use.
CitationLuís M. O. Matos, António J. R. Neves, Diogo Pratas and Armando J. Pinho. “MAFCO: a compression tool for MAF files”. PLoS ONE 10(3): e0116082.
DOI: http://dx.doi.org/10.1371/journal.pone.0116082.
DownloadACE’14 Workshop on “Designing Systems for Health and Entertainment: what are we missing?”
Systems that aggregate health and entertainment goals are proliferating, but little is known about the way to design and evaluate these systems and how to manage the different (if nor opposite) needs of these two main areas. This workshop will promote the discussion of issues surrounding these areas, enabling a better understanding of the how’s and why’s of designing systems for health and entertainment, as well as the identification of new avenues of research in the field.
Therefore we invite designers, researchers and practitioners to participate in an exciting full-day workshop where they are invited to share their personal views and research on the intersection of technology, health and entertainment.
More information at http://designingsystemsforhealthandentertainment.wordpress.com/.
SMBM 2014
The 6th International Symposium on Semantic Mining in Biomedicine (SMBM)
6th-7th October, 2014 will be held at the University of Aveiro, Portugal.
SMBM aims to bring together researchers from text and data mining in biomedicine, medical, bio- and chemoinformatics, and researchers from biomedical ontology design and engineering. SMBM 2014 is the follow-up event to SMBM 2012 (University of Zürich, Switzerland) SMBM 2010 (EBI, U.K.), SMBM 2008 (University of Turku, Finland), SMBM 2006 (University of Jena, Germany), and SMBM 2005 (EBI, U.K.).
More information at http://www.smbm.org.
PhD Defense (Luis Ribeiro)
Luis Ribeiro, “Platform for on-demand exchange of medical imaging communities”
Universidade de Aveiro, DETI/IEETA
PhD Defense (David Campos)
David Campos, “Term expansion methodologies in biomedical information retrieval”
Universidade de Aveiro, DETI/IEETA
Sérgio Matos was awarded a FCT Investigator grant
The FCT Investigator Programme aims to create a talent base of scientific leaders, by providing 5-year funding for the most talented and promising researchers, across all scientific areas and nationalities.
For the 2013 call, Sérgio Matos, research assistant at IEETA, was awarded a FCT Investigator grant, for the 2014-2018 period.
FALCON
 About
AboutFALCON is an alignment-free unsupervised system to measure a similarity top of multiple reads according to a database. The machine learning system can be used, for example, to classify metagenomic samples. The core of the method is based on the relative algorithmic entropy, a notion that uses model-freezing and exclusive information from a reference, allowing to use much lower computational resources. Moreover, it uses variable multi-threading, without multiplying the memory for each thread, being able to run efficiently from a powerful server to a common laptop. To measure the similarity, the system will build multiple finite-context (Markovian) models that at the end of the reference sequence will be kept frozen. The target reads will then be measured using a mixture of the frozen models. The mixture estimates the probabilities assuming dependency from model performance, and thus, it will allow to adapt the usage of the models according to the nature of the target sequence. Furthermore, it uses fault tolerant (substitution edits) Markovian models that bridge the gap between context sizes. Several running modes are available for different hardware and speed specifications. The system is able to automatically learn to measure similarity, whose properties are characteristics of the Artificial Intelligence field.
CitationPaper was submitted, currently the citation should be addressed to the url (bioinformatics.ua.pt/software/falcon).
DownloadDna-at-glance
 DNAatGlance is a program for the detection of large-scale genomic regularities by visual inspection. Several discovery strategies are possible, including the standalone analysis of single sequences, the comparative analysis of sequences from individuals from the same species, and the comparative analysis of sequences from different organisms. The software was designed and implemented at IEETA, a research unit of the University of Aveiro, and is available for non-commercial use.
DNAatGlance is a program for the detection of large-scale genomic regularities by visual inspection. Several discovery strategies are possible, including the standalone analysis of single sequences, the comparative analysis of sequences from individuals from the same species, and the comparative analysis of sequences from different organisms. The software was designed and implemented at IEETA, a research unit of the University of Aveiro, and is available for non-commercial use.
CitationArmando J. Pinho, Sara P. Garcia, Diogo Pratas, Paulo J. S. G. Ferreira (2013) DNA Sequences at a Glance. PLoS ONE 8(11): e79922.
DOI: dx.doi.org/10.1371/journal.pone.0079922.
DownloadFor convenience, we provide a sequence (here in gzip)(here in zip) and the corresponding information profile in WIG format (here in gzip) (here in zip) that can be uploaded to the UCSC Genome Browser as a custom track.
MFCompress
MFCompress: a compression tool for FASTA and multi-FASTA data
AboutMFCompress is a compression tool for FASTA and multi-FASTA files. In comparison to gzip and applied to multi-FASTA files, MFCompress can provide additional average compression gains of almost 50%, i.e., it potentially doubles the available storage, although at the cost of some more computation time. On highly redundant data sets, and in comparison with gzip, 8-fold size reductions have been obtained. MFCompress was designed and implemented at IEETA, a research unit of the University of Aveiro, and is available for non-commercial use. For other uses, please send an email to ap@ua.pt.
CitationArmando J. Pinho, and Diogo Pratas. “MFCompress: a compression tool for FASTA and multi-FASTA data.” Bioinformatics 30.1 (2014): 117-118.
DOI: dx.doi.org/10.1093/bioinformatics/btt594.
Downloadegas
Available at https://bioinformatics.ua.pt/egas.
What?
Egas is a web-based platform for biomedical text mining and collaborative curation. The web tool allows users to annotate texts with concept occurrences as well as with relations between concepts. Annotations can be performed manually or based on the results of automated concept identification and relation extraction tools. These automatic annotations may have been previously added to the documents, using one of the accepted input formats, or may be added during the annotation process, by calling a document annotation service. Users can inspect, correct or remove automatic text mining results, manually add new annotations, and export the results to standard formats.
How?
Text-processing and fetching modules, such as the concept and relation annotation services, were implemented in Java, and the web interface was developed using HTML5, CSS3, and JavaScript, in order to allow fast processing of large documents and support mobile devices. The resulting information is stored in a relational database. Finally, all database operations are performed using secured RESTful web-services, allowing easy integration with mobile devices, such as smartphones and tablets.
Cloud Thinking
Funding entity: QREN MaisCentro
Period: Feb.2013 – Dec.2014
The projects’ ambition is the creation of a new set of solutions based in novel ICT technologies, developing a concept that encompasses the synergistic usage of cloud computing, with large database access and information retrieval, associated with advanced methods for reasoning and data mining (and with the basic scalable algorithms to support the dimensions of the data sets targeted).
NeuroPath – New Strategies Applied to Neuropathological Disorders
Funding entity: QREN MaisCentro
Period: Feb.2013 – Jun.2015
Neurodegenerative disorders are a major health concern worldwide, Portugal being no exception. With this project the University of Aveiro proposes extend existing research in the field of neurodegenerative diseases through the creation of a consortium of 5 research units from UA (CBC, QOPNA, I3N, IEETA, CICECO). The projects main goal is to offer novel therapeutic strategies to tackle the complex array of existing neuropathologies. By building a multidisciplinary research team that combines experts in molecular neuropathologies, proteomics, metabolomics, bioinformatics, neuronal networks, organic synthesis and drug design from the UA we will be able to attack the problem on many fronts. Upon successful completion of this project, new therapeutic approaches will have been developed which will contribute to the improvement of life quality for neurodegenerative patients, having a high society impact considering the 10 million new patients reported every year.
Best Poster award at BioLINK SIG 2013
The price was awarded at BioLINK SIG 2013 for the work “Neji: a tool for heterogeneous biomedical concept identification”.
BioLINK SIG 2013: Roles for text mining in biomedical knowledge discovery and translational medicine
The Annual Meeting of the ISMB BioLINK Special Interest Group
In Association with ISMB/ECCB 2013, Berlin, Germany
July 20, 2013
iOS Development Seminar (Rui Pedro Lopes)
A 6-hour iOS Development Seminar will be held by Rui Pedro Lopes, Professor at Polytechnic Institute of Brangança, on the 29th July 2013, at Department of Electronics, Telecommunications and Informatics (DETI), Aveiro.
This Seminar will cover the following main topics: Objective-C, Storyboards, Core Data, Master-Detail User Interface
Universidade de Aveiro, DETI, Room 102, 10h
Variobox
 Exploring Human Genetic Variations
Exploring Human Genetic Variations
AboutVariobox is a desktop tool for the annotation, analysis and comparison of human genes. Variant annotation data are obtained from WAVe, protein metadata annotations are gathered from PDB and UniProt, and sequence metadata is obtained from Locus Reference Genomic (LRG) and RefSeq databases. By using an advanced sequence visualization interface, Variobox provides an agile navigation through the various genetic regions. Researched genes are compared to the sequences retrieved from LRG and RefSeq, automatically finding and annotating new potential mutations. These features and data, ranging from patient sequences to HGVS-valid variant description up to pathogenicity evaluation, are combined in an intuitive interface to explore genes and mutations.
CitingTo cite this tool use the following publication:
Variobox: Automatic Detection and Annotation of Human Genetic Variants. Paulo Gaspar, Pedro Lopes, Jorge Oliveira, Rosário Santos, Raymond Dalgleish, José Luís Oliveira. Human Mutation, 2014
DownloadVarioBox is available for all the main operating systems (Windows [XP, 7, 8]; Linux; MacOS) that support Java. The current version of the software is 1.4.4. Click the link bellow to download:
To run, first unpack all the files to any folder. Then, if you’re on Windows, double click the Variobox file inside the folder. On Mac or Linux, start a terminal, change the directory to the created folder, and run java -jar variobox.jar
Tutorial Step 1 The initial layout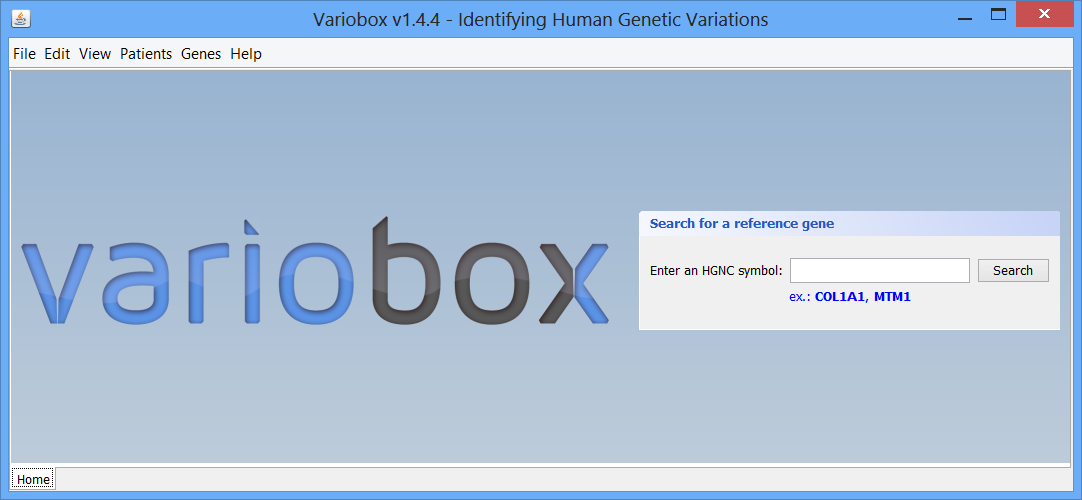 This is the initial VarioBox workspace that shows up when you open the application. At the bottom of the workspace you can find a separator, “Home”, created automatically. Here will be as many separators as searches performed, each one identified by the searched HGNC code. At the centre you can see the logo and a panel, where searches for reference genes can be performed, using a valid HGNC symbol. To work with Variobox, a reference gene is always the starting point. After obtaining the reference, a sequence can be loaded to the application to be aligned with the sequence, and analysed.
This is the initial VarioBox workspace that shows up when you open the application. At the bottom of the workspace you can find a separator, “Home”, created automatically. Here will be as many separators as searches performed, each one identified by the searched HGNC code. At the centre you can see the logo and a panel, where searches for reference genes can be performed, using a valid HGNC symbol. To work with Variobox, a reference gene is always the starting point. After obtaining the reference, a sequence can be loaded to the application to be aligned with the sequence, and analysed.
Step 2 Making a quick searchBy default there are two genes bellow the search box: Collagen, type I, alpha 1 (COL1A1), and Myotubularin 1 (MTM1). Click on COL1A1 or type it at the search box and hit search. A progress bar will show up indicating the progress of the loading process. A new tab (with the name of the searched HGNC code), like the one below, will show up once the reference gene is automatically retrieved from the web servers:
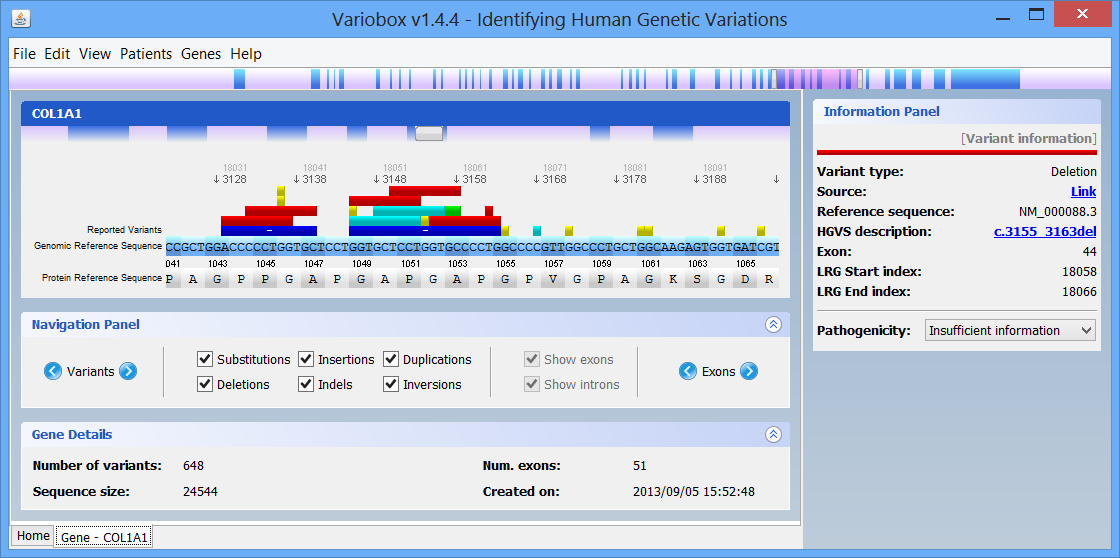
The right zone is formed by two distinct panels:
- The top one, titled Protein Viewer is where the 3D protein conformation of the selected gene is shown, if available, using JMol.
- The bottom one, titled Information Panel, which will display additional information on selected items, such as mutations and exons.
On the top of the window there is a large genomic viewer with a movable and resizable window that allows specifying a region to be explored in the centre zone. This viewer distinguishes exons (blue) and introns (purple), and allows quickly jumping through the gene. The centre zone is populated with gene data and information, in three distinct panels, described below:
- Gene panel
In this panel you can see the codon sequence and the decoded polypeptide sequence, labelled Reference Sequence and Translated Sequence respectively, and also the Known Mutations for the gene, as retrieved from WAVe. A zoomed genomic viewer is also displayed to further facilitate the exploration of the gene.
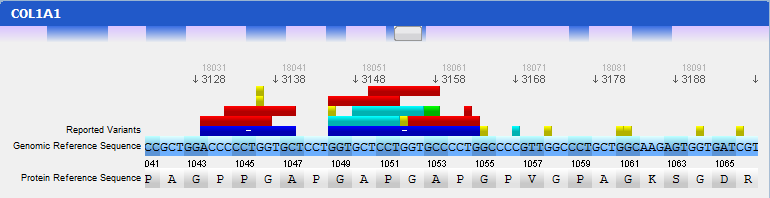
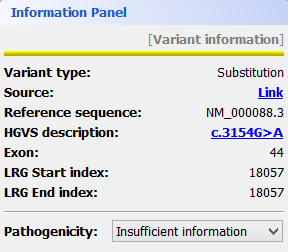
Mutations are identified by different colours, and shown next to the corresponding nucleotides. Additional information about a mutation can be obtain by clicking on the mutation. The Information Panel (right side of the workspace) will display details regarding the selected mutation’s position, source, type, annotation, etc.
- Navigation panel
The navigation panel is a simple feature that allows the easy exploration of the gene through mutations and exons. Clicking on the next or previous buttons will centre the sequence in the appropriate item (a mutation or exon):
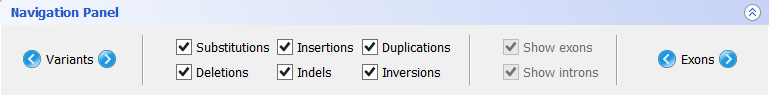
The Navigation Panel also permits filtering what mutation types are to be shown in the Gene Panel. For instance, if you only check Substitutions, all mutations besides SNPs will be hidden.
- Gene Details panel
This panel shows you a quick information about the gene that you are analysing. The current information supported is the following:
- Number of mutations: displays the total number of mutations found in the reference gene. No information will be displayed if no mutations are known;
- Number of exons: total number of exons found in the gene;
- Sequence size: total size of the reference sequence;
- Date of creation: the date and time when this gene was created;
- Loaded files: the files that were selected by the user to be aligned with the reference sequence.

Step 3 Loading mutated sequencesTo load a gene sequence and align it with the reference gene, click the menu Genes → Load gene file. Alternatively, go to the menu File → Load gene file. You will be prompted with a new window to select the file you want to load. For the current version we support the file types:
- DNA Sequence Chromatogram File: .scf ; .abi extension
- DNA Electropherogram File: .ab1 extension
- FASTA files: .fasta ; .fa extension
After selecting the file (or files, if you choose the forward-reverse format), click Load selected file and VarioBox will read them. Once the file is correctly loaded, an alignment with the reference gene is automatically performed. This alignment will also display found mutations, as compared to the reference gene. The analysis of the loaded sequence is described in the next step.
Step 4 Analysing mutated sequences and saving resultsAfter the files are loaded, the Gene Panel will be updated with the mutated sequence as well as the calculated mutations, as depicted in the following figure:
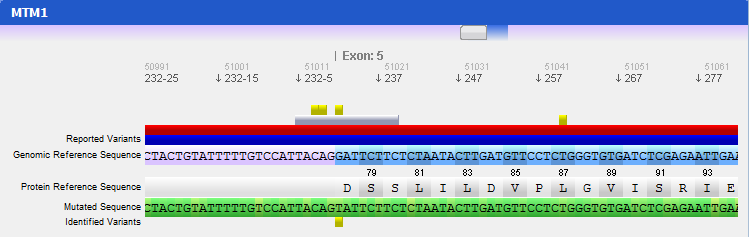
The loaded sequence will also be coloured according to its chromatogram confidence (if there is one), ranging from green (high confidence) to red (no confidence). This will allow easily understanding the validity of calculated mutations. Also note that the mutations are automatically annotated using the standard notation, and its annotation is displayed when clicking on a mutation. To save the sequences, mutations, alignment and other information, the gene should be assigned to a patient. To do so, go to the menu Genes → Save to patient and select a patient from the list of patients that will be presented.
Step 5 Final FeaturesIf you want to register a new patient in VarioBox, make the following steps: Go to Patients → New patient and fill the Patient Details panel (shown bellow) with all the required information(note that only one field is mandatory). After that just click Save patient and a new record will be created.
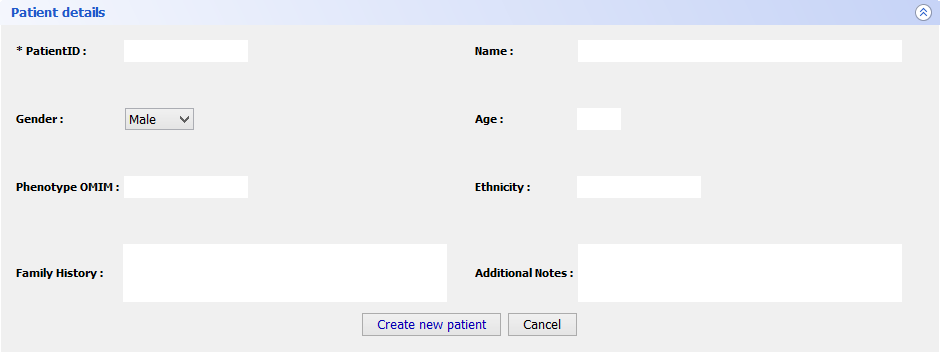
To load a saved project, go to Patients → Open patient and select the patient you previously saved. This will create a new tab with all the patient information: patient personal information as well as the genes from that patient. Those genes can be open just by selecting them and clicking Open selected.
This action will open many tabs as many genes you have selected and will re-create all the gene panels you had in the workspace previously.
Closing tabs is as simple as going to Patients → Close patient or Genes → Close current gene project depending of the tab type you have open.
becas

Biomedical Concept Annotation Tool, API and Widget
About
becas is a web application, API and widget for biomedical concept identification. It helps researchers, healthcare professionals and developers in the identification of over 1,200,000 biomedical concepts in text and PubMed abstracts.
becas provides annotations for isolated, nested and intersected entities. It identifies concepts from multiple semantic groups, providing preferred names and enriching them with references to public knowledge resources. You can choose the types of entities you want to identify and highlight or mute specific entities in real-time.
To facilitate annotation of PubMed abstracts, becas automatically fetches publications from NCBI servers and renders them with identified concepts highlighted.
Using becas
You can access the becas web annotation tool here and learn to use it in its help page. Explore the Web API in the API docs and discover how easy it is to integrate the becas widget in the widget docs.
You can read more about becas in the about page and we would love to hear your feedback!
Diseasecard
Diseasecard is a public web portal that integrates real-time information from distributed and heterogeneous medical and genomic databases, presenting it in a familiar visual paradigm.
Bioinformatics is playing a key role on molecular biology advances, not only by enabling new methods of research, but also managing the huge amounts of relevant information and make it available world-wide.
State of the art methods on bioinformatics include the use of public databases to publish the scientific breakthroughs. These databases provide valuable knowledge for the medical practice. But, given their specificity and heterogeneity, we cannot expect the medical practitioners to include their use in routine investigations. To obtain a real benefic from them, the clinician needs integrated views over the vast amount of knowledge sources, enabling a seamless querying and navigation.
Goals
Main goals behind the conception of DiseaseCard:
- Provide the user with an integrated view of the information available in the internet for a specific disease, from the phenotype to the genotype.
- Use rare diseases as the main target due to the high association between phenotype and genotype.
- Do not replicate information that already exists in public or private databases. The system is based in an information model that allows accessing and sharing these data;
- Be supported in a navigation protocol that allows guiding users in the process of retrieving information from the Internet.

DiseaseCard
Results
Diseasecard can provide the answers to several questions that are relevant in the genetic diseases diagnostic, treatment and accomplishment, such as:
- What are the main features of the disease?
- Are there any drugs for the disease?
- Are there any gene therapies for the disease?
- What laboratories perform genetic tests for the disease?
- What genes cause the disease?
- On which chromosomes are these genes located?
- What mutations have been found in these genes?
- What names are used to refer to these genes?
- What are the proteins coded by these genes?
- What are the functions of the gene product?
- What is the 3D structure for these proteins?
- What are the enzymes associated to these proteins?
Publications
- G. Dias, J. L. Oliveira, F. Vicente, and F. Martín-Sanchez, “Integrating Medical and Genomic Data: a Sucessful Example for Rare Diseases”, in The XX International Congress of the European Federation for Medical Informatics (MIE’2006), Maastricht, Netherlands, 2006.
- G. Dias, J. L. Oliveira, F. Vicente, and F. Martin-Sanchez, “Integration of Genetic and Medical Information Through a Web Crawler System”, in Biological and Medical Data Analysis (ISBMDA’ 2005), Lecture Notes in Computer Science – Volume 3745, Aveiro, Portugal, 2005.
nccd
 About
AboutNCCD is a method and package tool designed to compute the NCCD (Normalized Conditional Compression Distance) and, for instance, to perform phylogenomics (whole genome) on 48 bird species. It will use a state-of-the-art genomic compressor, based on a mixture of finite-context models, as a metric distance.
CitationD. Pratas, A. J. Pinho. “A conditional compression distance that unveils insights of the genomic evolution.” arXiv preprint arXiv:1401.4134 (2014).
DownloadEU-ADR Web Platform

The EU-ADR Web Platform helps experts in the study of adverse drug reactions (ADRs) through the use of computational services and scientific workflows, provided by several European partners. The system assists in the earlier detection of adverse drug reactions, improving drug safety and contributing to public health benefit. You can access the EU-ADR Web Platform here
EU-ADR Project
The overall objective of this project was the design, development and validation of a computerized system that exploits data from electronic healthcare records and biomedical databases for the early detection of adverse drug reactions. Visit the project page.
On Multicriteria Pairwise Sequence Alignment: Algorithms and Applications
Talk from Luís Paquete, Anf. IEETA
The multiobjective formulation of the pairwise sequence alignment problem is introduced, where a vector score function takes into account the substitution score and indels or gaps separately. Two solution methods are introduced: a multiobjective dynamic programming that extends classical algorithms for this problem and an epsilon-constraint algorithm that solves a series of constrained sequence alignment problems. A state pruning technique based on the concept of bound sets is also presented. Finally, its application to phylogenetic tree construction is
discussed.
Universidade de Aveiro, Anf. IEETA, 14h30
EMIF – European Medical Information Framework
Funding entity: IMI-JU
Period: 2013-2018
In recent years, the development and use of Electronic Healthcare Records (EHRs) throughout Europe has grown exponentially resulting in large volumes of clinical data. At the same time, large collections of disease‐specific data are recorded – in local, regional and/or national settings. Researchers also follow specific cohorts over time, and focus on specific types of data such as imaging or genetic data. Other researchers are building biobanks that aim to combine clinical data with genetic data. As a result, individual patients can contribute to multiple, often separate, data sources.
RD-CONNECT – An integrated platform connecting registries, biobanks and clinical bioinformatics for rare disease research
Funding entity: FP7-HEALTH-2012-INNOVATION-1
Period: 2012-2018
Despite examples of excellent practice, rare disease (RD) research is still mainly fragmented by data and disease types. Individual efforts have little interoperability and almost no systematic connection between detailed clinical and genetic information, biomaterial availability or research/trial datasets. By developing robust mechanisms and standards for linking and exploiting these data, RD-Connect will develop a critical mass for harmonisation and provide a strong impetus for a global “trial-ready” infrastructure ready to support the IRDiRC goals for diagnostics and therapies for RD patients.
Neji
![]()
Flexible, easy and powerfull framework for faster biomedical concept recognition.
Download Learn more
What?
Neji is an innovative framework for biomedical concept recognition. It is open source and built around four key characteristics: modularity, scalability, speed, and usability. It integrates modules of various state-of-the-art methods for biomedical natural language processing (e.g., sentence splitting, tokenization, lemmatization, part-of-speech tagging, chunking and dependency parsing) and concept recognition (e.g., dictionaries and machine learning). The most popular input and output formats, such as Pubmed XML, IeXML, CoNLL and A1, are also supported. Additionally, the recognized concepts are stored in an innovative concept tree, supporting nested and intersected concepts with multiples identifiers. Such structure provides enriched concept information and gives users the power to decide the best behavior for their specific goals, using the included methods for handling and processing the tree.
Why?
Concept recognition is an essential task in biomedical information extraction, presenting several complex and unsolved challenges. The development of such solutions is typically performed in an ad-hoc manner or using general information extraction frameworks, which are not optimized for the biomedical domain and normally require the integration of complex external libraries and/or the development of custom tools. Thus, Neji fills the gap between general frameworks (e.g., UIMA and GATE) and more specialized tools (e.g., NER and normalization), streamlining and facilitating complex biomedical concept recognition.
How?
On top of the built-in functionalities, developers and researchers can implement new processing modules or pipelines, or use the provided command-line interface tool to build their own solutions, applying the most appropriate techniques to identify names of various biomedical entities. Neji was built thinking on different development configurations and environments: a) as the core framework to support all developed tasks; b) as an API to integrate in your favorite development framework; and c) as a concept recognizer, storing the results in an external resource, and then using your favorite framework for subsequent tasks.
Systems Biology seminars series start on the 28th of September
Universidade de Aveiro, Anf. Ambiente, 14h
PhD Defense (Pedro Lopes)
Pedro Lopes, “Service Composition in Biomedical Applications”
Universidade de Aveiro, DETI/IEETA
Talk (Kim Sneppen)
Dr. Kim Sneppen from the Niels Bohr Institute, Copenhagen-DK, will give the give the inaugural Lecture of our Systems Biology seminars series entitled Simplified Models of Biological Networks, on the 28th of September.
Universidade de Aveiro, Anf. Ambiente, 14h
mRNA Optimiser
 Redesign mRNA sequences to optimise the secondary structure
Redesign mRNA sequences to optimise the secondary structure
AboutThe mRNA optimiser is a tool that redesigns a gene messenger RNA to optimise its secondary structure, without affecting the polypeptide sequence. The tool can either maximize or minimize the molecule minimum free energy (MFE), thus resulting in decreased or increased secondary structure strength.
The optimisation is achieved by using an heuristic to look for synonymous gene sequences, and select the ones with the best secondary structure. Evaluations of the secondary structure are made using a correlated stem-loop prediction algorithm that examines the nucleotide sequence for simple stem-loops. This algorithm is fine-tuned to have its results highly correlated with the MFE evaluations of RNAfold.
Our results indicate that an average of over 40% increase in MFE can be obtained with this method. Also, since there is a tendency to reduce the GC percentage of nucleotide sequences when optimising, the developed tool includes an option to maintain the GC content of the wildtype gene.
CitingP. Gaspar, G. Moura, M. A. S. Santos, and J. L. Oliveira mRNA secondary structure optimization using a correlated stem–loop prediction Nucleic Acids Research, Jan 2013, doi: 10.1093/nar/gks1473
DownloadSelect your operating system:



Current version is 1.0.
UsageThe mRNA optimiser is a command line tool (a graphical interface will be available soon). To use it you need to open a terminal window, change to the directory where mRNAOptimiser is, and run it:
1. Open a terminal window
- In Windows, go to the Start menu, click Run, write cmd, and click Ok.
- In Mac, write terminal in spotlight and hit enter.
2. Change the directory
- In Windows, Mac and Linux, write cd in the terminal followed by the directory where you placed the tool.
3. Run the mRNA optimiser
- In Windows, write mRNAOptimizer.exe and hit enter. Usage indications will show up in the terminal.
- In Mac and Linux, write java -jar mRNAOptimizer.jar and hit enter. Usage indications will show up in the terminal.
You may choose to supply your mRNA sequence by writing it into the terminal or referring an input file, with the -f input_sequence option. The tool only changes the coding region of the mRNA, therefore you must indicate where the start codon begins (-b index, to indicate the index of the first nucleotide of the start codon) and where the stop codon ends (-e index, to indicate the index of the last nucleotide of the stop codon). The default coding zone is the entire sequence.
To redirect the output results to a file, use the -o output_file option. To choose whether the tool should maximize or minimize the MFE, use the -d type option (default is maximize). You may limit the algorithm in both time and number of iterations by using the options -t max_time and -i max_iterations. Also, the tool will use the standard genetic code by default, but you can select other genetic coding tables using the -c coding_table option.
To maintain the original mRNA percentage of guanine and citosine (GC content) unaltered after optimisation, use the -g option. There is also a quiet mode, where nothing is output except for the resulting sequence, using the -q option.
Any questions and suggestions are welcome 🙂
OralCard
What is OralCard?
OralCard is an online bioinformatic tool that comprises results from manually curated articles reflecting the oral molecular ecosystem (OralPhysiOme), by merging the experimental information available from the oral proteome both of human (OralOme) and microbial origin (MicroOralOme). OralCard is a key resource for understanding the molecular foundations implicated in biology and disease mechanisms of the oral cavity.
How does it work?
OralCard integrates information about more than 3500 proteins and searching can be performed in three distinct views: (1) by protein names or respective UniProt codes, (2) by disease name, OMIM code or MeSH term, (3) and by organism.
PhD Defense (Nuno Rosa)
Nuno Rosa, “From the Salivar Proteome to Oralome”
Universidade Católica Portuguesa, Viseu
Eugene
International School on SWAT4LS 2012 (May 2nd – 5th, Aveiro)
International School on Semantic Web Applications and Technologies for the Life Sciences 2012
May 2nd – 5th, 2012
Located at the University of Aveiro,
Aveiro, Portugal
More information online at http://www.swat4ls.org/schools/aveiro2012/
Talk (Helena Deus)
Helena Deus, “Linked Data and Semantic Web Technologies for improving discovery in the Life Sciences”
We live in a world of data. This is also true for the Life Sciences, where the introduction of omics technologies such as genome sequencing has led to the industrialization of data production beyond a craft-based cottage industry and into a deluge of biological information. Nevertheless, the apparently simple task of collecting and keeping pace with the latest information about a gene of interest is still thwarted by the need for biological researchers to become experts at database-surfing and literature mining.
Linked Data is a set of principles devised for creating a Web of Data where a new generation of Web applications can discover and link relevant pieces of information based on its properties rather than its location in a database. Linked data is also at the root of a movement towards building a knowledge continuum in the Life Sciences and by doing so, has the potential to be a foundation for a platform that will support 21st century Biology.
In this talk, I will present some of the scenarios where Linked Data has been successfully applied in accelerating scientific discovery and translation of Life Sciences knowledge into Health Care and what challenges are still to be addressed.
Helena Deus Bio at http://lenadeus.info
GReEn

GReEn: a tool for efficient compression of genome resequencing data.
GReEn: a tool for efficient compression of genome resequencing data.
AboutResearch in the genomic sciences is confronted with the volume of sequencing and resequencing data increasing at a higher pace than that of data storage and communication resources, shifting a significant part of research budgets from the sequencing component of a project to the computational one. Hence, being able to efficiently store sequencing and resequencing data is a problem of paramount importance.
We describe GReEn (Genome Resequencing Encoding), a tool for compressing genome resequencing data using a reference genome sequence. It overcomes some drawbacks of the recently proposed tool GRS, namely, the possibility of compressing sequences that cannot be handled by GRS, faster running times and compression gains of over 100-fold for some sequences.
GReEn is available for non-commercial use. For other uses, please send an email to ap@ua.pt.
We describe GReEn (Genome Resequencing Encoding), a tool for compressing genome resequencing data using a reference genome sequence. It overcomes some drawbacks of the recently proposed tool GRS, namely, the possibility of compressing sequences that cannot be handled by GRS, faster running times and compression gains of over 100-fold for some sequences.
GReEn is available for non-commercial use. For other uses, please send an email to ap@ua.pt.
Download Citation
Armando J. Pinho, Diogo Pratas, Sara P. Garcia. (2012). GReEn: a tool for efficient compression of genome resequencing data. Nucleic acids research, 40(4), e27-e27.
DOI: 10.1093/nar/gkr1124
DOI: 10.1093/nar/gkr1124
Best PhD work in the Fraunhofer Portugal Challenge 2011

III Workshop Ibero-NBIC – 2011
III WORKSHOP DE RED IBEROAMERICANA DE TECNOLOGÍAS CONVERGENTES NBIC EN SALUD (IBERO-NBIC) – CYTED Program
Hotel Moliceiro, Aveiro, Portugal
October 10-11, 2011
Day 1: Monday, 10
9h00 – 9h30: Opening and Welcome
Boas Vindas
José Luís Oliveira, DETI/IEETA, Universidade de Aveiro, Portugal
Acto de apertura del III Workshop Internacional Redes Ibero-NBIC y NanoRoadmap
Alejandro Pazos & Julián Dorado, Universidade da Coruña, España
9h30 – 11h15:
Vacunología inversa aplicada en malaria
Raúl Isea, IDEA, Fundación de Estudios Avanzados, Venezuela
Bioinformatics, research and applications
Sergio Guíñez Molinos, UCBSM, Universidad de Talca, Chile
Tecnologías NBIC y Nanotoxicidad: Gestión del conocimiento asociado al uso de nanopartículas en medicina
Diana de la Iglesia, GIB, Universidad Politécnica de Madrid, España
Tecnologías de la Información y el Conocimiento en Salud. Un Sistema Basado en Ontologías para el Apoyo a la Toma de Decisión en UCIs
Ana Freire, Universidade de Coruña, España
11h15 – 11h30: Coffee Break
11h30 – 13h00:
Integration of heterogeneous biomedical names taggers
David Campos, DETI/IEETA, Universidade de Aveiro, Portugal
Collecting and Enriching Human Variome Datasets
Pedro Lopes, DETI/IEETA, Universidade de Aveiro, Portugal
Doctoral Program in Nanosciences and Nanotechnology of the University of Aveiro
Tito Trindade, DQ, Universidade de Aveiro, Portugal
13h00 – 14h30: Lunch
14h30 – 16h00:
Integración de la información molecular en un Sistema de Informacion en Salud
Segunda etapa: estándares y control de calidad.
Carlos Otero, HIBA, Buenos Aires, Argentina
Connecting different levels of biological information. From atoms to people
Guillermo López, ISCIII – Instituto de Salud Carlos II, Madrid, España
Posibles aportes de una empresa de Educación Médica Continua a una red de investigación en Salud
Antonio López, EVIMED, Uruguay
16h00 – 16h30: Coffee Break
16h30 – 18h00:
Internal Meeting / Reunión Interna de la Red
Day 2: Tuesday, 11
Visit to Instituto Ibérico de Nanotecnologia (Braga)
Gimli

Annotation of biomedical entity names
the best open-source solution
 Open Source
Open Source- Use, change and distribute
- Social development
 High Performance*
High Performance*- BioCreative: 87,54%
- JNLPBA: 73,05%
 High-end Techniques
High-end Techniques- Linguistic dependency parsing
- Model combination
 Flexible and Scalable
Flexible and Scalable- Extensible architecture
- Fast annotation
 Easy to Use
Easy to Use- Automated scripts
- Java library
 License
License
- Non-commercial use
* Overall F-measure results achieved using the evaluation methods of the respective challenges.
About
Goal:
Gimli is a machine learning-based solution for biomedical Named Entity Recognition (NER), which goal is to automatically extract names of biomedical entities from scientific text documents. Currently, Gimli supports the recognition of gene/protein, DNA, RNA, cell line and cell type names.
In summary, Gimli receives raw text as input, and provides text with specific annotations as output.
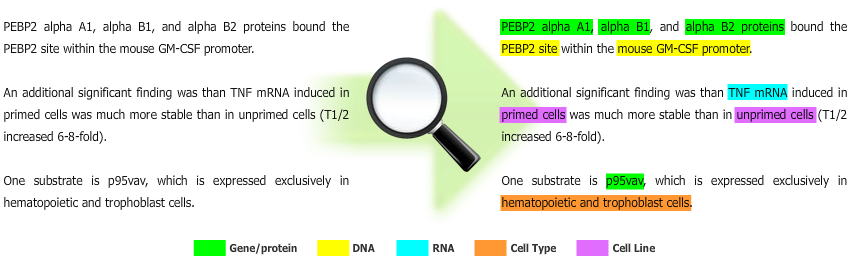
Methods:
- Machine Learning: Conditional Random Fields (CRFs);
- Features: orthographic, morphological, linguistic parsing and conjunctions;
- Combination: combination of models with different orders and parsing directions;
- Post-processing: parentheses correction and abbreviation resolution.
Publication(s):
- David Campos, Sérgio Matos, José Luís Oliveira. Gimli: open source and high-performance biomedical name recognition. BMC Bioinformatics, vol. 14, no. 1, p. 54, February 2013
Download
Tool:
Get the latest official release of Gimli.
Source code:
Get a copy of the project using the following git command:
 git clone git://github.com/davidcampos/gimli.git
git clone git://github.com/davidcampos/gimli.gitDocumentation
Full documentation:
Complete information about alternative downloads, installation and usage.
API Javadoc:
Detailed classes, methods and propreties description.
Join us
We have several ideas to make Gimli the most complete and efficient tool for biomedical information extraction. You are welcome to join us and contribute to the development of new and improved features. Please contact us:
 david.campos(a)ua.pt
david.campos(a)ua.ptTeam
- David Campos, david.campos(at)ua.pt
- Sérgio Matos, aleixomatos(at)ua.pt
- José Luís Oliveira, jlo(at)ua.pt
Totum

Problem
The recognition of named entities is a crucial initial task of biomedical text mining. A number of NER solutions have been proposed in recent years, taking advantage of different resources and/or techniques. Currently, the best results are achieved by combining the output of different systems. However, little effort has been spent in such harmonisation solutions, being specific to a corpus and/or non-knowledge based.
Features
Conceptual
- Knowledge-based harmonisation
- Correct, remove and create annotations
- Support several biomedical domains and organisms
- On-demand harmonisation
- Support both NER and normalisation systems
Technical
- Automated scripts for simple usage
- Java library for advanced users
- Input and Output in IeXML format
Method
Totum is a innovative harmonisation solution based on Conditional Random Fields, which were trained on several manually curated corpora. Thus, we avoid the single corpus dependency, supporting several biomedical domains and organisms. In the end, Totum harmonises gene/protein annotations provided by several heterogeneous NER solutions, following the gold standard requirements.

Results
Considering a corpus that contains the test parts of the four corpora, the experiments show that Totum improves the F-measure of state-of-the-art tagging solutions by up to 10% in exact alignment and 22% in nested alignment. Finally, Totum achieves an F-measure of 70% (exact matching) and 82% (nested matching) against the same corpus.
Used tools
- MALLET: framework for statistical natural language processing, providing a Conditional Random Fields implementation;
- Apache OpenNLP: tokenisation and respective model;
- IeXML: annotation guidelines and associated library;
- monq.jfa: fast and flexible text filtering with regular expressions.
Publication(s)
- David Campos, Sérgio Matos, Ian Lewin, José Luís Oliveira, Dietrich Rebholz-Schuhmann. Harmonisation of gene/protein annotations: towards a gold standard MEDLINE. Bioinformatics, vol. 28, no. 9, p. 1253-1261, March 2012. doi:10.1093/bioinformatics/bts125
Team
Partners
Members
- David Campos, david.campos(at)ua.pt
- Sérgio Matos, aleixomatos(at)ua.pt
- Ian Lewin, lewin(at)ebi.ac.uk
- José Luís Oliveira, jlo(at)ua.pt
- Dietrich Rebholz-Schuhmann, rebholz(at)ebi.ac.uk
COEUS
COEUS main web server is down for maintenance. It will be online again on February 27th, 2013. Thank you for your patience.
Ipsa scientia potestas est. Knowledge itself is power.
Streamlined back-end framework for rapid semantic web application development.
Get it Here
GitHub project

Integration
Create custom warehouses, integrating distributed and heterogeneous data.
Integrate CSV, SQL, XML or SPARQL resources with advanced Extract-Transform-Load warehousing features.

Cloud-based
Deploy your knowledgebase in the cloud, using any available host.
Your content – available any time, any where. And with full create, read, update, and delete support.

Semantics
Use Semantic Web & LinkedData technologies in all application layers.
Enable reasoning and inference over connected knowledge.
Access data through with LinkedData interfaces and deliver a custom SPARQL endpoint.

Rapid Dev Time
Reduce development time. Get new applications up and running much faster using the latest rapid application development strategies.
COEUS is the back-end framework, the client-side is language-agnostic: PHP, Ruby, JavaScript, C#… COEUS’ API works everywhere.

Interoperability
Use COEUS advanced API to connect multiple nodes together and with any other software.
Create your own knowledge network using SPARQL Federation enabling data-sharing amongst a scalable number of peers

Ecosystem
Launch your custom application ecosystem. Distribute your data to any platform or device.
Reach more users and create new semantic cloud-based software platforms.
XGB 2011 Best poster award
WAVe
The Human Variome relates to genomic mutations and their effects on particular phenotypes. This critical life sciences research field has grown greatly in recent years, mostly due to the appearance of projects such as the Human Variome Project or the European GEN2PHEN Project. Nonetheless, locus-specific mutation databases and included variants are far from being standardized and widely used in the research community workflow. With WAVe, we offer centralized and transparent access to these databases, combined with the integration of found variants in a single system that is enriched with the most relevant gene-related information in a user-friendly web-based workspace.
https://bioinformatics.ua.pt/WAVe
Features
WAVe provides a comprehensive set of features that will improve bioligists’ workflow when researching in the genomic variation field.
Search
Searching for genes only requires that users start typing the gene HGNC-approved symbol in any of the available search boxes. This event will trigger the automatic suggestion system that will offer various solutions based on users’ input. Following one of the suggestions leads directly to the gene view interface. When a suggestion is not accepted and there is more than one match, WAVe will display the gene browse interface, containing only the results matching the provided query.
Browse
Querying for * lists all genes as well as available LSDBs and variants for each gene. In this gene browse scenario, searches for a particular gene can be performed, in real time, by typing in the table search box. By clicking in one of the genes, users are sent to the gene view interface.
View
The gene view interface is the main WAVe workspace. The layout is divided in two main areas: the sidebar and the content zone. The sidebar displays minimal gene information on top – gene HGNC symbol, name and locus – and the navigation tree, which is WAVe’s user interface key element, at the bottom. The navigation tree is organized in nodes, each referring to a distinct data type: each node leaf links directly to a page containing information regarding a specific topic. Pages linked in each leaf appear in the content zone. This enables loading external applications without leaving WAVe’s interface and, thus, without losing focus with ongoing research.
API
Programmatic access to data is also available. The gene tree is available as an easily-parsable feed. Feeds are obtained by appending the atom tag (or other format: rss, json) to the end of the gene view address. For instance, BRCA2 Atom feed is available at https://bioinformatics.ua.pt/WAVe/gene/BRCA2/atom .
WAVe also provides an RSS API for variant access. With this, you have programmable access to all available variants for a given gene. For instance, BRCA2 variants (from multiple LSDBs) are at https://bioinformatics.ua.pt/wave/variant/BRCA2/atom. In addition to the variant description, WAve points to the original LSDB containing the variant.
This WAVe makes WAVe the only platform capable of providing aggregated variant listings through both visual and programmable access.
Feedback
We highly appreciate any feedback you can provide regarding WAVe and the genomic variation field. To do this, you can simply send an e-mail to pedrolopes@ua.pt. Thank you.
Talk (Daniel Sobral)
Daniel Sobral, “Ensembl Regulation”
Ensembl is a world reference for vertebrate genome annotation, providing high quality annotation for more than 50 species. Particularly challenging is the annotation of non-coding functional regions of the genome. Ensembl Regulation aims at making Ensembl
a reference for the annotation of genomic features with a potential role in the transcriptional regulation of gene expression. Combining publicly available data from large projects like ENCODE and The Epigenomics Roadmap, we group overlapping areas of open chromatin and transcription factor binding to build a “best-guess” set of regulatory features, in a cell-aware manner. Finally, we also include histone-modification and polymerase data to generate cell-specific classifications for the regulatory regions. Taking advantage of the role of the EBI as part of the ENCODE data analysis group, we aim at bringing Ensembl to the forefront of the annotation of the regulatory genome.
PhD Defense (Daniel Polónia)
Daniel Polónia, “An electronic market for teleradiology services”
PhD Defense (José Paulo Lousado)
José Paulo Lousado, “Pattern analysis on DNA primary structure”
EU-ADR – Early Detection of Adverse Drug Events by Integrative Mining of Clinical Records and Biomedical Knowledge
Funding entity: FP7-ICT (STREP)
Period: 2008-2012
The overall objective of this project is the design, development and validation of a computerized system that exploits data from electronic healthcare records and biomedical databases for the early detection of adverse drug reactions.
GeNS
Genomic Name Server
The integration of heterogeneous data sources has been a fundamental problem in database research over the last two decades. The goal is to achieve better methods to combine data residing at different sources, under different schemas and with different formats in order to provide the user with a unified view of the data. Although simple in principle, due to several constrains, this is a very challenging task where both the academic and the commercial communities have been working and proposing several solutions that span a wide range of fields. However, the limitations found on most solutions reflect the difficulty to obtain a simple but comprehensive schema able to accommodate the heterogeneity of the biological domain while maintaining an acceptable level of performance: GeNS is our proposal towards solving this issue.
Installing and using GeNS
The Genomic Name Server can be either downloaded and installed on a local computer or accessed by Web Services. Please keep in mind that GeNS currently requires over 10 GB of disk space and this figure is likely to increase in the near future. Therefore, if disk space is a serious restriction you should consider using the available Web Services. We are currently using
Microsoft SQL Server 2008 but GeNS can be set up in any other DBMS.
a) Setting up a local instance of GeNS
- Download either the full backup of the database (here) or a dump of all the tables (available here): Last update: 24/11/09
- Once inside your DMBS, simply restore the full backup of the database (this is for MS SQL Server 2008 only; a step-by-step walkthrough can be found here) or import the data from the tables to the database.
- Congratulations! GeNS is now ready to be used.
b) Using the Web Services
The Web Services are now available here. Furthermore, a detailed description is also available here (Updated March 24).The Web Services API is in an early stage of development and, as such, users should bear in mind that certains problems may arise during it’s usage.
Advantages
- Easy to understand and use
- Flexible and scalable
- Efficient
- Accessible by several methods
- Improves the cross-database low identifier coverage issue
Architecture
GeNS uses four distinct methods for gathering data from external databases: by Web Services, web crawlers, database connectors and finally by tabular files connectors. All of the recovered data is subsquently processed and synchronized to our database. Finally, the data can be accessed via Web Services or by downloading, installing and querying the data with SQL.
Currently, GeNS is importing data from four major databases: UniProt (SwissProt and TrEMBL), KEGG, EMBL – EBI and Entrez. Since these databases already incorporate data from third-party databases, we have over 460.000 unique genes, more than 100.000 biological relations and a hundred and forty distinct datatypes.

Architecture
Database
GeNS database was designed with simplicity and extensibility in mind; the following schema is a complete representation of the database.

Database
Concepts:
- Organism: An individual form of life capable of growing, metabolizing nutrients, and usually reproducing. Organisms can be unicellular or multicellular. The Organism table stores taxonomic information; each entry corresponds to an organism with any given number of associated proteins. This table is the root of the hierarchical model. For each organism, we store its scientific and short names.
- Protein: Any of a group of complex organic macromolecules that contain carbon, hydrogen, oxygen, nitrogen, and usually sulfur and are composed of one or more chains of amino acids. The Protein table is where the proteins’ internal identifiers and gene locus are stored; each entry in this table has a referring organism (in which this protein is found) and may have any number of associated biological entities and/or equivalent external databases’ protein identifiers in the ProteinIdentifier and BioEntity tables.
- ProteinIdentifier: The table in which the mapping between the external databases’ protein identifier and BioPortal’s
internal identifier is made. - BioEntity: A table that stores all the biological entities associated with a given protein; this includes,
among other things, pathways and gene ontologies. - DataType: A table listing all the possible external databases from which the biological data may come from; each entry in the ProteinIdentifier and BioEntity tables references this
table, so that we may easily determine the nature (and source) of the
data.
Reproducing the results
The following files allow anyone to reproduce the obtained results regarding the cross-database low identifier coverage issue and the
performance testing queries. You will need a working copy of GeNS in order to use these scripts.
GeneBrowser
GeneBrowser is a web-based tool that, for a given list of genes, combines data from several public databases with visualisation and analysis methods to help identify the most relevant and common biological characteristics. The functionalities provided include the following: a central point with the most relevant biological information for each inserted gene; a list of the most related papers in PubMed and gene expression studies in ArrayExpress; and an extended approach to functional analysis applied to Gene Ontology, homologies, gene chromosomal localisation and pathways.

GeneBrowser
Although GeneBrowser can be used to answer many different biological questions, a particular question set was used to tune its development:
- What public databases provide relevant information about my dataset and how can I navigate through them?
- What biological processes are enriched with respect to my input list of genes?
- What are the most relevant metabolic pathways that contain my genes?
- What are the genomic regions of these genes?
- Which are the most relevant homologue classes in my list of genes?
- What gene expression experiments have been previously conducted with the same genes?
- What are the most relevant publications associated with my study?
Feedback
We highly appreciate any feedback you can provide regarding GeneBrowser. jpa@ua.pt. Thank you.
Reference
J. Arrais, J. Fernandes, J. Pereira and J. L. Oliveira, Exploring and identifying common biological traits in a set of genes, BMC Bioinformatics, BMC Bioinformatics 2010, 11:212 (link)
Dicoogle
Dicoogle is an open-source Picture Archiving and Communications System (PACS) archive. Its modular architecture allows the quick development of new functionalities, due to the availability of a Software Development Kit (SDK).
Dicoogle starts by indexing DICOM files and metadata, both locally and in distributed systems using a P2P communication framework. Upon this distributed index users can then search for exams or specific features using a free text interface.
QuExT
What is QuExT?
QuExT (Query Expansion Tool) is a document indexing and retrieval application that obtains, from the MEDLINE database, a ranked list of publications that are most significant to a particular set of genes. Document retrieval and ranking are based on a concept-based methodology that broadens the resulting set of documents to include documents focusing on these gene-related concepts. Each gene in the input list is expanded to its various synonyms and to a network of biologically associated terms. Currently, the expansion is based on proteins, metabolic pathways and diseases (this last one only when the selected organism is Homo sapiens). The retrieved documents are ranked according to user-definable weights for each of these concept classes. By simply changing these weights, users can alter the order of the documents, allowing them to obtain for example, documents that are more focused on the metabolic pathways in which the initial genes are involved, rather than on the genes themselves.
How does it work?
QuExT receives as input a list of genes and a corresponding organism. The gene list can be typed into the input box or uploaded in a text file. Genes can be separated by commas or spaces. The organism to consider is selected from the drop-box menu. Figure 1 shows the query expansion procedure.
When the user submits the form, gene names or identifiers in the input are checked against a database and mapped to an internal identifier corresponding to the selected organism. Genes which are not found in the database are rejected from further analysis.
QuExT then creates an expanded query and searches a local index of the PubMed database for documents matching this query.
Query expansion is performed as follows: for each gene in the query, the algorithm obtains, from a term expansion table corresponding to the selected organism, all the alternative gene, protein, pathway and disease names corresponding to that gene’s internal ID. The full list of terms from all input genes is then accumulated in four separate query strings (one for each concept type). Each term obtained from expanding all genes is used to search the index.
QuExT runs four index searches using the four query strings obtained in the query expansion stage (one for each concept type). For each search, the documents that match the query and the corresponding scores are obtained. Resulting documents and corresponding scores are kept on separate lists, one for each concept class.
Notice that while the term expansion takes into account the selected organism, to avoid going from a gene in one organism to a related term in a different organism, this is not true for document retrieval. Since the indexing does not distinguish between different species referred in the articles, a search for a gene name in H. sapiens may return results referring to the same gene but in mice, for example.
Finally, the results from the document retrieval stage are assembled and documents are re-ranked in terms of the defined weights for each concept. The final score for document i is obtained as a weighted sum of the four concept-based scores:

where Wj is the weight attributed to the concept type j and sij represents the score for document i in terms of the jth concept type.
References
S. Matos, J. P. Arrais, J. Maia-Rodrigues, J. L. Oliveira, “Concept-based query expansion for retrieving gene related publications from MEDLINE”, BMC Bioinformatics, Apr 28; 11:212, 2010.
Neoscreen
NeoScreen is a bioinformatics software that helps diagnosis tasks in newborn screening programs. The application imports MS/MS raw data, and organizes and maintains all the information along the time in a database, providing a set of patterns that allow the detection of abnormalities in the blood samples. Is is been used, from 2005, to support the Portuguese Newborn Screning Program (http://www.diagnosticoprecoce.org/)
NeoScreen – Newborn screening analysis
The introduction of the Tandem Mass Spectrometry (MS/MS) in neonatal screening laboratories has opened the way to innovative newborn screening analysis. With this technology the number of metabolic disorders that can be detected, from dried blood-spot species, increases significantly. However, the amount of information obtained with this technique and the pressure for quick and accurate diagnostics raises serious difficulties in the daily data analysis. To face this challenge we developed a software system, NeoScreen, which simplifies and allow speeding up newborn screening diagnostics.
Software
In this view, the individuals are separated in several diagnostic categories, such as “very suspicious”, “suspicious”, “not suspicious”, etc. Some of these categories represent individuals with markers out of the established limits, but that are not associated with any known disease. In the right-side frame it is displayed the relevant information that was extracted and processed by the software for each individual, like: plate information, markers concentrations, and suspicious diseases.

Neoscreen
Since May 2011, NeoScreen is represented by BMD Software Lda.
Mind
MIND is a repository of microarray experiments that handles storage, management and analysis of microarray data. It is supported by an infrastructure prepared to integrate dynamically further functionalities (Quality Control assurance, data processing, data mining, visualization, reports, etc.).
Microarray INformation Database
The development of microarray technology has been phenomenal during the past years, and it is becoming a daily tool in many genomics research laboratories. However, the multi-step and data-intensive nature of this technology has created an unprecedented computational challenge. In fact, the full power of microarrays technology can only be achieved if researchers are able to efficiently store, analyse and share their results.

MIND Workflow
LIMS capabilities
A LIMS (Laboratory Information Management System) is an database repository that allows to manage all the laboratorial data.

MIND LIMS
Main advantages of MIND:
- Easier and fast access to all the laboratorial data
- Trace of all the experiment allowing errors detection
- Allows an easier share of data among different users
- Public web-based interface
- MIAME and MAGE compliance
Data Analysis capabilities

MIND Data Analysis
Quality control
- Enables the user to detect systematic errors on the production of microarrays. It also allows the usage of some pre-processing such as background subtraction, data normalization and data filtering;
Exploratory data analysis
- Allows the user to, based on definite objectives, specify the experiment design and retrieve the biological meaning from the shown results.
Software integration
- Allow the dynamic introduction of processing algorithms and R scripts.
Publications
- J. Arrais, J. L. Oliveira, G. Grimes, S. Moodie, K. Robertson, and P. Ghazal, “Microarray data sharing in BioMedicine”, in The XX International Congress of the European Federation for Medical Informatics (MIE’2006), Maastricht, Netherlands, 2006.
- J. Arrais, L. Carreto, M. A. S. Santos, and J. L. Oliveira, “Collaborative work on microarrays using MAGE-ML”, in 9th International Meeting of the Microarray Gene Expression Data Society (MGED9), Seattle, Washington, USA, 2006.
Anaconda
ANACONDA is a software package specially developed for the study of genes’ primary structure. It uses gene sequences downloaded from public databases, as FASTA and GenBank, and it applies a set of statistical and visualization methods in different ways, to reveal information about codon context, codon usage, nucleotide repeats within open reading frames (ORFeome) and others.
Codon context analysis
Genome sequencing is opening unprecedent ways for understanding how gene primary structure is organized. Two of the most studied open reading frame characteristics are codon usage and codon context.
Traditional methods used for codon usage and context analysis do not provide user-friendly tools to carry out detailed gene primary structure analysis at a genomic scale.
Codon usage tables, using absolute metric, are available in public databases for any sequenced gene or genome and freeware software for multivariate analysis (correspondence analysis) of codon and amino acid usage is also readily available, however sophisticated statistical and data visualization tools are clearly lacking.
We propose the usage of several statistical methods – contingency table analysis, residual analysis, multivariate analysis (cluster analysis) – to analyze the codon bias under various aspects (degree of association, contexts and clustering).
Cluster analysis
A cluster analysis tool allows also calculating similarities between two vectors of the contingency table. This technique is used to group lines and columns (codons) of the correlation matrix, allowing highlight global patterns in the genes.
The statistical tools that are incorporated in the system, for data clustering, residual analysis and histogram plotting of calculated indexes, allow reaching new conclusions on gene primary structure features at a genomic scale. We expect that the results obtained will permit identifying some general rules that govern codon context and codon usage in any genome. Additionally, the identification of genes containing expanded codons that arise as a consequence of erroneous DNA replications events will permit uncovering new genes associated to human disease.
Visualization
In order to detect the impact of codon context bias (as well as the presence of rare codons) on coding sequences, ANACONDA has additional tools for sequence mapping. The layout for sequence include written information about the ORF and the sequence itself, in which the codons have been coloured with the same residual colour scale of the ORFeome map.
ANACONDA allows the user to work with more than one ORFeome at a time. This creates large data sets that are difficult to deal with, in particular when multiple comparisons are being performed.
Considering that vast number of ORFeomes can be analyzed simultaneously by ANACONDA, we have included extra tools to allow comparative studies.

Anaconda
he statistical tools that are incorporated in the system, for data clustering, residual analysis and histogram plotting of calculated indexes, allow reaching new conclusions on gene primary structure features at a genomic scale. We expect that the results obtained will permit identifying some general rules that govern codon context and codon usage in any genome.
Past publications
- G. Moura, M. Pinheiro, J. Arrais, A. C. Gomes, L. Carreto, A. Freitas, J. L. Oliveira, and M. A. Santos, “Large Scale Comparative Codon-Pair Context Analysis Unveils General Rules that Fine-Tune Evolution of mRNA Primary Structure”, PLoS ONE, vol. 2, no. 9, e847, doi:10.1371/journal.pone.0000847, 2007.
- M. Pinheiro, V. Afreixo, G. Moura, A. Freitas, M. A. Santos, and J. L. Oliveira, “Statistical, computational and visualization methodologies to unveil gene primary structure features”, Methods of Information in Medicine, vol. 45, no. 2, pp. 163-168, 2006.
- G. Moura, M. Pinheiro, R. Silva, I. Miranda, V. Afreixo, G. Dias, A. Freitas, J. L. Oliveira, and M. A. Santos, “Comparative context analysis of codon pairs on an ORFeome scale”, Genome Biology, vol. 6, no. 3, pp. R28, 2005.
Download
Anaconda 2 is now available for download. It is freely available for fundamental research only.
[Download]
Last Update (2011-01-12)
New features (Version 2.0, 2011):
- Corrected some bugs
- Enriched codon statistics and visualization maps
- Comparing context maps across several species
- Integration of tRNA copy number processing
- Single and multiple sequence alignments using codon context (BLASTP and ClustalW)
Himage PACS
A PACS solution for echocardiography laboratories that provides a cost-efficient digital archive, and enables the acquisition, storage, transmission and visualization of DICOM cardiovascular ultrasound sequences.

Scenario
The medical imaging digitalization and implementation of PACS (Picture Archiving and Communication Systems) systems increases practitioner’s satisfaction through improved faster and ubiquitous access to image data. Besides, it reduces the logistic costs associated to the storage and management of image data and also increases the intra and inter institutional data portability. Echocardiography is a rather demanding medical imaging modality when regarded as digital source of visual information. The date rate and volume associated with a typical study poses several problems. They are hard to keep “online” (in centralized servers) and difficult to access (in real-time) outside the institutional broadband network infra-structure. For example, an uncompressed echocardiography study size can typically vary between 100 and 500Mbytes.
Product Presentation
The innovation of our approach is the implementation of a DICOM private transfer syntax designed to support any video encoder installed on the operating system. This structure provides great flexibility concerning the selection of an encoder that best suits the specifics of a particular imaging modality or working scenario. To ultrasound studies we are using the highly efficient MPEG4 codec that takes full advantage of object texture, shape coding and inter-frame redundancy. More than 40.000 studies have been performed so far. For example, a typical Doopler color run (RGB) with an optimized time-acquisition (15-30 frames) and a sampling matrix (480*512), rarely exceed 200-300kB. Typical compression ratios can go from 65 for a single cardiac cycle sequence to 100 in multi- cycle sequences. With these averaged figures, even for a heavy work-loaded echolab, it is possible to have all historic procedures online or distribute them with reduced transfer time over the network, which is a very critical issue when dealing with costly or low bandwidth connections. The solution is actually installed in one public Central Hospital (CHVNG) and one private laboratory of cardiac images. Because the solution front-end is fully Web-based, the clinical specialists are using the platform to provide decision support remotely, accessing over Internet in a secure way (i.e. over SSL). Moreover, the solution is changing the work methods. The process workflow is fully digital where reviewing and reporting procedures can be done at physician’s home (i.e. telework).

- MS-PDC
- MS-PDC

Himage Modules
- Visualization – view dicom medical images
- Report Module – edit and export a image report, with customizable layout, to Word.
- Burning Module – to export the study to CD/DVD in DICOM default transfer syntax, including a standalone viewer.
- Communications Module – send a study to a external server.
Himage Modules
Image Quality
Two studies were carried on assessing the DICOM cardiovascular ultrasound image quality. In a simultaneous and blind display of the original against the compressed cine-loops, 37% of the trials have selected the compressed sequence as the best image. This suggests that other factors related with viewing conditions are more likely to influence observer performance than the image compression itself.
Developing new tools for studying mRNA mistranslation
Funding entity: FCT (PTDC/BIA-BCM/72251/2006)
Period: 2008-2011
Implementation of a Nacional Facility for DNA Microarrays: Phase II
Funding entity: FCT (PTDC/BIA-BCM/64745/2006)
Period: 2009-2011
New statistical methodologies for analysis DNA microarrays data
Funding entity: FCT (PTDC/MAT/72974/2006)
Period: 2006-2008
DNA Microarray technology is one of the most promising new technologies for global gene expression analysis. This technology is sophisticated, very expensive, highly interdisciplinary and produces vast amounts of data whose management and analysis pose significant challenges. This project aims to study new bi-clustering approaches that can help to obtain relevant information from gene expression microarrays.
mRNA mistranslation in yeast
Funding entity: HFSP Research Grant
Period: 2005-2008
The very few quantitative mRNA mistranslation studies carried out to date indicate that the average decoding error ranges from 10-4 to 10-5 errors per codon decoded. However, no systematic study has yet been carried out to rank mRNA sequences according to
decoding error and no methodology has yet been developed to identify genes that are prone to decoding error.
In this project, software tools for data visualization and mathematical methodologies for identification of general rules governing RNA translation, and tools for mapping mRNA regions of high decoding error and for identifying putative gene expression regulatory sequences present in mRNAs, will be developed.
INFOBIOMED – Structuring European Biomedical Informatics to Support Individualised Healthcare
Funding entity: IST FP6 (IST2002-507585) – NoE (Network of Excelence)
Period: 2003-2006
There is a great potential for synergy between medical informatics and bioinformatics with a view on continuity and individualisation of healthcare, so that the benefits of the human genome sequence can reach the population. A collaborative effort between those two disciplines is needed to bridge the current gap between them. Biomedical Informatics (BMI) is an emerging discipline that aims at bringing these two worlds together to foster the development of novel diagnostic and therapeutic methodologies and strategies.
The INFOBIOMED network aims at setting a durable structure for the described collaborative approach at an European level, mobilising the critical mass and the resources necessary for enabling the collaborative approach that supports the consolidation of BMI as a crucial scientific discipline for future healthcare.
(http://www.infobiomed.org/)
INFOGENMED – A virtual laboratory for accessing and integrating genetic and medical information for health applications
Funding entity: IST FP5 (IST2001-39013)
Period: 2002-2004
UA/IEETA was the Project Coordinator
One goal currently challenging bio – and clinical informatics is to develop robust computational methods and tools to model, store, retrieve and analyse information at multiple levels of complexity, i.e., from molecule to organism. For example, the unification of heterogeneous databases under one virtual system is an important step towards developing such robust computational models. The latter is the objective of the INFOGENMED project which aims at building a virtual laboratory for accessing and integrating genetic and medical information for health applications. Once built, the system allows practitioners, biologists, chemists and other experts to navigate through local and remote biomedical databases.
INFOGENMED started in September 2002, (http://infogenmed.web.ua.pt/), and the functionalities already built in the system allow for: (1) defining clinical pathways to guide the user in the navigation of multiple sources over the Internet; (2) identifying and characterizing the most relevant databases to support the molecular medicine practice for selected rare genetic diseases; (3) designing the integration methods, based on virtual databases, mediators and semantic vocabulary servers.
Next Generation Information Systems
Talk from Florentino Fernández Riverola, Dpto. de Informática – Universidade de Vigo
Current research lines and projects of the “Next Generation Information Systems” group, from University of Vigo, in Orense
14th Annual Meeting of the Portuguese Human Genetics Society
A 14ª Reunião da Sociedade Portuguesa de Genética Humana irá realizar-se nos dias 18, 19 e 20 de Novembro de 2010, em Coimbra, no Auditório da Fundação Bissaya Barreto (Bencanta).
Mais informação na página da SPGH.
ITAB2010
The 10th IEEE International Conference on Information Technology and Applications in Biomedicine,
will be held in Corfu, Greece, November 2-5, 2010 at Aquis Corfu Holiday Palace.
X Jornadas Bioinformática 2010
The Xth Spanish Symposium on Bioinformatics (JBI2010) take place in October 27-29, 2010 in Torremolinos-Málaga, Spain. Co-organised by the National Institute of Bioinformatics-Spain and the Portuguese Bioinformatics Network and hosted by the University of Malaga (Spain).
23rd tRNA Workshop, Jan 2010
Place: Aveiro, Portugal
Date: 28 Jan- 2 Feb 2010
XV National Congress of Biochemistry (NCB2006)
Place: Aveiro, Portugal
Date: December 8-10, 2006
IV International Symposium on Biological and Medical Data Analysis (ISBMDA 2005)
Place: Aveiro, Portugal
Date: November 10-11, 2005
Books and Chapters
Books
- O. Bodenreider, J.L Oliveira, and F. Rinaldi (Eds.)
Proceedings of the 6th International Symposium on Semantic Mining in Biomedicine (SMBM 2014), University of Aveiro, 2014 - M. Santos (Ed.)
XVth National Congress of Biochemistry, 2006
ISBN 978-972-789-214-3 - M. Santos (Ed.)
IIIrd National Meeting on RNA Biology, 2006
ISBN 978-972-789-215-0 - J. L. Oliveira, V. Maojo, F. Martin-Sanchez, and A. S. Pereira (Eds.),
Biological and Medical Data Analysis, Springer, 2005,
ISBN 978-972-789-215-0, http://www.springerlink.com/content/k15u6315h070/
Book chapters
- O. Fajarda, A. Trifan, M. Van Speybroeck, P. R. Rijnbeek, J. L. Oliveira
“Exploring the Value of Electronic Health Records from Multiple Datasets”
in Biomedical Engineering Systems and Technologies. BIOSTEC 2018. Communications in Computer and Information Science , vol 1024, Cliquet Jr. A. et al. (eds), Springer, Cham, 2019.pdf - D. Canedo, A. Trifan, A. J. R. Neves
“Monitoring Students’ Attention in a Classroom Through Computer Vision”
in Communications in Computer and Information Science, J. Bajo, J. M. Corchado et al., Eds., Springer International Publishing, 2018.pdf - C. V. Ferreira and C. Costa
“Challenges of Using Cloud Computing in Medical Imaging”
in Advances in Cloud Computing Research, M. Ramachandran, 2014. - J. Melo, J. P. Arrais, E.Coelho, P. Lopes, N. Rosa, M. J. Correia, M.Barros and J. L. Oliveira
“Data Integration Solution for Organ-Specific Studies: An Application for Oral Biology”
in Biomedical Engineering Systems and Technologies, J. Gabriel, J. Schier, S. Huffel et al, Eds., Springer Berlin Heidelberg, 2013. - A. Freitas, W. Ayadi, M. Elloumi, J. L. Oliveira, and J.-K. Hao
“A Survey on Biclustering of Gene Expression Data”
in Biological Knowledge Discovery Handbook: Preprocessing, Mining and Postprocessing of Biological Data, M. Elloumi & A. Zomaya, Eds., Wiley, 2013. - D. Campos, S. Matos, and J. L. Oliveira
“Current methodologies for biomedical Named Entity Recognition”
in Biological Knowledge Discovery Handbook: Preprocessing, Mining and Postprocessing of Biological Data, M. Elloumi & A. Zomaya, Eds., Wiley, 2013. - M. Santos, L. Bastião, C. Costa, A. Silva, N. Rocha
“Clinical Data Mining in Small Hospital PACS: Contributions for Radiology Department Improvement”
in Information Systems and Technologies for Enhancing Health and Social Care, IGI Global, 2013. - F. Valente, C. Costa, and A. Silva
“Content Based Retrieval Systems in a Clinical Context”
in Medical Imaging in Clinical Practice, Okechukwu F. Erondu, Ed., InTech, February 2013. - D. Campos, S. Matos, and J. L. Oliveira
“Biomedical Named Entity Recognition: a Survey of Machine-Learning Tools”
in Theory and Applications for Advanced Text Mining, S. Sakurai, Ed., InTech, 2012. - P. Lopes and J. L. Oliveira
“Collecting and Enriching Human Variome Datasets”
in Tecnologías NBIC en Salud: El papel protagonista de la Nanociencia, J. Aguiló, A. Freire, D. Iglesia, V López, A Pazos, Eds., CYTED, 2012, pp. 9-19. - L. S. Ribeiro, C. Costa and J. L. Oliveira
“Current Trends in Archiving and Transmission of Medical Images”
in Medical Imaging, Okechukwu F. Erondu, Ed., InTech, December 2011. - J. Arrais, S. Matos, and J. L. Oliveira
“Integração de Dados Biomédicos”
in Nano, Bio, Info y Cogno (Convergencia de Tecnologías NBIC) Conceptos y Aplicaciones, J Aguiló, A Freire, D Iglesia, F Martin, A Pazos, Eds., 2011, pp. 185-226. - G. Moura, M. Pinheiro, A. Freitas, J. L. Oliveira, and M. A. Santos
“Computational and Statistical Methodologies for ORFeome Primary Structure Analysis”
in Comparative Genomics, Methods in Molecular Biology series, N. Bergman, Ed.: Humana Press, USA, 2007, pp. 449-462. - C. Costa, A. Silva, and J. L. Oliveira
“Current Perspectives on PACS and a Cardiology Case Study”
in Advanced Computational Intelligence Paradigms in Healthcare 2, vol. 65, S. Vaidya, L. C. Jain, and H. Yoshida, Eds.: Springer-Verlag, 2007, pp. 79-108. - C. Costa, A. Silva, J. L. Oliveira, V. Ribeiro, and J. Ribeiro
“A demanding Web-based PACS supported by Web Services technology”
in Medical Imaging 2006: PACS and Imaging Informatics, vol. 6145, O. R. Steven C.Horii, Ed., 2006. - C. Costa, J. L. Oliveira, A. Silva, V. Ribeiro, and J. Ribeiro
“Himage: Um Web-PACS inovador para Imagem Cardíaca”
in TIC em Biomedicina, vol. 6145, M. G. J. R. Rabunal, N. Pedreira, J. Pereira, Ed. Santiago de Compostela 2005.
Journals
- A. Pereira, R. P. Lopes and J. L. Olivera
“SCALEUS-FD: A FAIR Data Tool for Biomedical Applications”
BioMed Research International, volume 2020, 2020 pdf - R. Lebre, L. B. Silva and C. Costa
“A Cloud-ready Architecture for Shared Medical Imaging Repository”
Journal of Digital Imaging, p. 1-12, 2020 pdf - J. R. Almeida, A. J. Pinho, J. L. Oliveira, O. Fajarda, D. Pratas
“GTO: A toolkit to unify pipelines in genomic and proteomic research”
SoftwareX, volume 12, p. 100535, 2020 pdf - O. Fajarda, S. Pereira, R. Silva, J. L. Oliveira
“Merging microarray studies to identify a common gene expression signature to several structural heart diseases”
BioData Mining, volume 13, no. 8, p. 1-20, 2020 pdf - M. Pedrosa, A. Zúquete, C. Costa
“RAIAP: renewable authentication on isolated anonymous profiles”
Peer-to-Peer Networking and Applications, p. 1-23, 2020 pdf - D. Pratas, M. Hosseini, J.M. Silva, A.J. Pinho
“A reference-free lossless compression algorithm for DNA sequences using a competitive prediction of two classes of weighted models”
Entropy, volume 31, no. 11, p. 1074, 2019 pdf - T.Godinho, R. Lebre, J.R. Almeida and C. Costa
“ETL Framework for Real-Time Business Intelligence over Medical Imaging Repositories”
Journal of Digital Imaging, volume 32, no. 5, p. 870-879, 2019 pdf - R. Antunes and S. Matos
“Extraction of chemical–protein interactions from the literature using neural networks and narrow instance representation”
Database, volume 2019, Article ID baz095, 2019 pdf - F.B. Correia, E. D. Coelho, J. L. Oliveira and J. P. Arrais
“Handling noise in protein interaction networks”
BioMed Research International, volume 2019, Article ID 8984248, 13 pages, 2019 pdf - A. Trifan, M. Oliveira and J. L. Oliveira
“Passive Sensing of Health Outcomes Through Smartphones: Systematic Review of Current Solutions and Possible Limitations”
JMIR mHealth and uHealth, volume 7, no. 8 p. e12649, 2019 pdf - J. R. Almeida, R. Gini, G. Roberto, P. Rijnbeek and J. L. Oliveira
“TASKA: A modular task management system to support health research studies”
BMC medical informatics and decision making, volume 19, no. 1 p. 121, 2019 pdf - A. Trifan and J. L. Oliveira
“Patient data discovery platforms as enablers of biomedical and translational research: a systematic review”
Journal of Biomedical Informatics, volume 93, p. 103154, 2019 pdf - J. M. Silva, T. Godinho, D. Silva and C. Costa
“A community-driven validation service for standard medical imaging objects”
Computer Standards & Interfaces, volume 61, p. 121-128, 2019 pdf - J. L. Oliveira, A. Trifan and L. B. Silva
“EMIF Catalogue: A collaborative platform for sharing and reusing biomedical data”
International Journal of Medical Informatics, volume 126, p. 35-45, 2019 pdf - R. I. Doğan, S. Kim, A. Chatr-aryamontri, C.-H. Wei, D. C. Comeau, R. Antunes, S. Matos, Q. Chen, A. Elangovan, N. C. Panyam, K. Verspoor, H. Liu, Y. Wang, Z. Liu, B. Altınel, Z. M. Hüsünbeyi, A. Özgür, A. Fergadis, C.-K. Wang, H.-J. Dai, T. Tran, R. Kavuluru, L. Luo, A. Steppi, J. Zhang, J. Qu and Z. Lu
“Overview of the BioCreative VI Precision Medicine Track: mining protein interactions and mutations for precision medicine”
Database (Oxford), volume 2019, no. bay147, 2019 pdf - M. Cavalheiro, C. Costa, A. Silva-Dias, I. M. Miranda, C. Wang, P. Pais, S. N. Pinto, D. Mil-Homens, M. Sato-Okamoto, A. Takahashi-Nakaguchi, R. M. Silva, N. P. Mira, A. M. Fialho, H. Chibana, A. G. Rodrigues, G. Butler and M. C. Teixeira
“Unveiling the mechanisms of in vitro evolution towards fluconazole resistance of a Candida glabrata clinical isolate: a transcriptomics approach”
Antimicrobial Agents and Chemotherapy, volume 63, no. 1, 2019, pdf - S. Matos
“Configurable web-services for biomedical document annotation”
Journal of Cheminformatics, volume 10, p. 68, 2018, pdf - M. Pedrosa, J. M. Silva, J. F. Silva, S. Matos, C. Costa
“SCREEN-DR: Collaborative platform for diabetic retinopathy”
International Journal of Medical Informatics, volume 120, p. 137-146, 2018 pdf - E. Pinho and C. Costa
“Automated Anatomic Labeling Architecture for Content Discovery in Medical Imaging Repositories”
Journal of Medical Systems, volume 42, no. 145, 2018, pdf - E. Pinho and C. Costa
“Unsupervised Learning for Concept Detection in Medical Images: A Comparative Analysis”
Applied Sciences, volume 8, no. 1213, 2018, pdf - A.F. Pedrosa, C. Lisboa, I. Faria-Ramos, R.M. Silva, I. M. Miranda and A. G. Rodrigues
“Malassezia species retrieved from skin with pityriasis versicolor, seborrheic dermatitis and skin free of lesions: a comparison of two sampling methods”
British Journal of Dermatology, volume 179, no. 2, p. 526-527, 2018, pdf - J. M. Silva, E. Pinho, E. Monteiro, J. F. Silva and C. Costa
“Controlled searching in reversibly de-identified medical imaging archives”
Journal of Biomedical Informatics, volume 77, p. 81–90, 2018, pdf - J. M. Silva, T. M. Godinho, D. Silva and C. Costa
“A Community-Driven Validation Service for Standard Medical Imaging Objects”
Computer Standards & Interfaces, 2018, pdf - L. B. Silva, A. Trifan and J. L. Oliveira
“MONTRA: An agile architecture for data publishing and discovery”
Computer Methods and Programs in Biomedicine, volume 160, p. 33-42, 2018, pdf - S. Matos and R. Antunes
“Protein-Protein Interaction Article Classification Using a Convolutional Recurrent Neural Network with Pre-trained Word Embeddings”
Journal of Integrative Bioinformatics, vol. 14, no. 4, December 2017, pdf - R. Antunes and S. Matos
“Supervised Learning and Knowledge-Based Approaches Applied to Biomedical Word Sense Disambiguation”
Journal of Integrative Bioinformatics, vol. 14, no. 4, December 2017, pdf - P. Sernadela and J. L. Oliveira
“COEUS 2.0: An automated platform to integrate and publish biomedical data as nano publications”
IET Software, 2017, pdf - A. Syzdykova, A. Malta, M. Zolfo, E. Diro, J. L. Oliveira
“Open-Source Electronic Health Record Systems for Low-Resource Settings: Systematic Review”
JMIR Medical Informatics, vol. 5, no. 4, Nov. 2017, pdf - P. Sernadela, L. González-Castro, C. Carta, E. van der Horst, P. Lopes, R. Kaliyaperumal, M. Thompson, R. Thompson, N. Queralt-Rosinach, E. Lopez, L. Wood, A. Robertson, C. Lamanna, M. Gilling, M. Orth, R. Merino-Martinez, M. Posada, D. Taruscio, H. Lochmuller, P. Robinson, M. Roos, and J. L. Oliveira
“Linked Registries: Connecting Rare Diseases Patient Registries through a Semantic Web Layer”
Biomed Research International, vol. 2017, pp. 1–13, Oct. 2017, pdf - J. Branco, M. Ola, R. Silva, E. Fonseca, N.C. Gomes, C. Martins-Cruz, A.P. Silva, A. Silva-Dias, C. Pina-Vaz, C. Erraught, L. Brennan, A.G. Rodrigues, G. Butler, I.M. Miranda
“Impact of ERG3 mutations and expression of ergosterol genes controlled by UPC2 and NDT80 in Candida parapsilosis azole resistance”
Clinical Microbiology and Infection, vol. 23(8), 575, Aug. 2017, pdf
- J. M. Silva, T.M. Godinho, D. Silva and C. Costa, C
“Web Validation Service for Ensuring Adherence to the DICOM Standard”
Studies in Health Technology and Informatics, volume 235, p.38–42, 2017, pdf - T. M. Godinho, R. Lebre, L. Silva and C. Costa
“An efficient architecture to support digital pathology in standard medical imaging repositories”
Journal of Biomedical Informatics, vol. 71, p. 190-197, July 2017, pdf - T.M. Godinho, C. Costa and J.L. Oliveira
“Intelligent generator of big data medical imaging repositories”
IET Software, vol. 11(3) July 2017, pdf - E. Monteiro, C. Costa and J.L. Oliveira
“A de-identification pipeline for ultrasound medical images in DICOM format”
Journal of Medical Systems, vol. 41(5), pp. 89, May 2017, pdf - P. Sernadela, L. González-Castro, and J. L. Oliveira
“SCALEUS: Semantic Web Services Integration for Biomedical Applications”
Journal of Medical Systems, vol. 41(4), pp.54, Apr. 2017, pdf - A.H.M.P. Tavares, A.J. Pinho, R. Silva, J.M.O.S. Rodrigues, C.A.C. Bastos, P.J.S.G. Ferreira, V. Afreixo
“DNA word analysis based on the distribution of the distances between symmetric words”
Scientific Reports, vol. 7, 728, Apr. 2017, pdf - V. Afreixo, J.M.O.S. Rodrigues, C.A.C. Bastos, A.H.M.P. Tavares, R.M. Silva
“Exceptional Symmetry by Genomic Word: A Statistical Analysis”
Interdisciplinary Sciences: Computational Life Sciences, vol. 9(1), pp. 14-23, Mar. 2017, pdf - E. Pinho, T. Godinho, F. Valente, C. Costa
“A Multimodal Search Engine for Medical Imaging Studies”
Journal of Digital Imaging, vol. 30(1), pp. 39–48, Feb. 2017, pdf - P. Sernadela, J. L. Oliveira, “A semantic-based workflow for biomedical literature annotation”
Database, Volume 2017, 1 Jan. 2017, pdf - L. Azevedo, M. Mort, A.C. Costa, R.M. Silva, D. Quelhas, A. Amorim, D.N. Cooper
“Improving the in silico assessment of pathogenicity for compensated variants”
European Journal of Human Genetics, vol. 25(1) Jan. 2017, pdf - F. Pereira, S. Duarte-Pereira, R.M. Silva, L.T. da Costa, I. Pereira-Castro
“Evolution of the NET (NocA, Nlz, Elbow, TLP-1) protein family in metazoans: insights from expression data and phylogenetic analysis”
Scientific Reports, vol. 6, 38383, Dec 2016, pdf - A. Marote, N. Barroca, R. Vitorino, R.M. Silva, M.H.V. Fernandes, P.M. Vilarinho, O.A.B. da Cruz e Silva, S.I. Vieira
“A proteomic analysis of the interactions between poly(L-Lactic Acid) nanofibers and SH-SY5Y neuronal-like cells”
AIMS Molecular Science, vol. 3(4), pp. 661-682 Nov. 2016, pdf - S. Kim, R. I. Doğan, A. Chatr-Aryamontri, C. S. Chang, R. Oughtred, J. Rust, R. Batista-Navarro, J. Carter, S. Ananiadou, S. Matos, A. Santos, D. Campos, J. L. Oliveira, O. Singh, J. Jonnagaddala, H.-J. Dai, E. C.-Y. Su, Y.-C. Chang, Y.-C. Su, C.-H. Chu, C. C. Chen, W.-L. Hsu, Y. Peng, C. Arighi, C. H. Wu, K. Vijay-Shanker, F. Aydın, Z. M. Hüsünbeyi, A. Özgür, S.-Y. Shin, D. Kwon, K. Dolinski, M. Tyers, W. J. Wilbur, D. C. Comeau
“BioCreative V BioC track overview: collaborative biocurator assistant task for BioGRID”
Database (Oxford), vol. 2016, September 2016, pdf - Q. Wang, S. S. Abdul, L. Almeida, S. Ananiadou, Y. I. Balderas-Martínez, R. Batista-Navarro, D. Campos, L. Chilton, H.-J. Chou, G. Contreras, L. Cooper, H.-J. Dai, B. Ferrell, J. Fluck, S. Gama-Castro, N. George, G. Gkoutos, A. K. Irin, L. J. Jensen, S. Jimenez, T. R. Jue, I. Keseler, S. Madan, S. Matos, P. McQuilton, M. Milacic, M. Mort, J. Natarajan, E. Pafilis, E. Pereira, S. Rao, F. Rinaldi, K. Rothfels, D. Salgado, R. Silva, O. Singh, R. Stefancsik, C.-H. Su, S. Subramani, H. D. Tadepally, L. Tsaprouni, N. Vasilevsky, X. Wang, A. Chatr-Aryamontri, S. J. F. Laulederkind, S. Matis-Mitchell, J. McEntyre, S. Orchard, S. Pundir, R. Rodriguez-Esteban, K. Van Auken, Z. Lu, M. Schaeffer, C. H. Wu, L. Hirschman, C. N. Arighi
“Overview of the interactive task in BioCreative V”
Database (Oxford), vol. 2016, September 2016, pdf - S. Matos, D. Campos, R. Pinho, R. M. Silva, M. Mort, D. N. Cooper, J. L. Oliveira
“Mining clinical attributes of genomic variants through assisted literature curation in Egas”
Database (Oxford), vol. 2016, June 2016, pdf - J. Arevalo, F. A. González, R. Ramos-Póllan, J. L. Oliveira, M. Guevara
“Representation learning for mammography mass lesion classification with convolutional neural networks”
Computer Methods and Programs in Biomedicine, p. 248–257, April 2016, pdf - E. Monteiro, C. Costa, and J. L. Oliveira
“A Cloud Architecture for Teleradiology-as-a-Service”
Methods of Information in Medicine, March 2016, pdf - P. Sernadela, P. Lopes, J. L. Oliveira
“A knowledge federation architecture for rare disease patient registries and biobanks”
Journal of Information Systems Engineering & Management, vol.2, March 2016, pdf - C. Viana-Ferreira, L. Ribeiro, S. Matos, C. Costa
“Pattern Recognition for Cache Management in Distributed Medical Imaging Environments”
International Journal of Computer Assisted Radiology and Surgery, vol. 11, no. 2, p. 327-336, February 2016. pdf - V. Afreixo, S. J. M. O. Rodrigues, C. C. A. Bastos, and M. R. Silva
“The exceptional genomic word symmetry along DNA sequences”
BMC Bioinformatics, vol. 17, pp. 1-10, February 2016. pdf - S. Duarte-Pereira, I. Pereira-Castro, S. S. Silva, M. G. Correia, C. Neto, L. T. da Costa, et al.
“Extensive regulation of nicotinate phosphoribosyltransferase (NAPRT) expression in human tissues and tumors”
Oncotarget, December 2015. pdf - A. R. Soares, N. Fernandes, M. Reverendo, H. R. Araújo, J. L. Oliveira, G. M. R. Moura, et al.
“Conserved and highly expressed tRNA derived fragments in zebrafish”
BMC Molecular Biology, vol. 16, pp. 1-16, December 2015. pdf - J. Branco, A. P. Silva, R. M. Silva, A. Silva-Dias, C. Pina-Vaz, G. Butler, et al.
“Fluconazole and Voriconazole Resistance in Candida parapsilosis Is Conferred by Gain-of-Function Mutations in MRR1 Transcription Factor Gene”
Antimicrob Agents Chemother, vol. 59, pp. 6629-33, October 2015. pdf - F. Valente, L. B. Silva, T. Godinho, C. Costa
“Anatomy of an Extensible Open Source PACS”
Journal of Digital Imaging, pp. 1-13, Ocotober 2015, pdf - P. Lopes and J. L. Oliveira
“An automated real-time integration and interoperability framework for bioinformatics”
BMC Bioinformatics, October 2015, pdf - P. Lopes, L. Bastiao Silva, and J. L. Oliveira
“Challenges and Opportunities for Exploring Patient-Level Data”
BioMed Research International, August 2015, pdf - R. M. Silva, D. Pratas, L. Castro, A. J. Pinho, and P. J. S. G. Ferreira
“Three minimal sequences found in Ebola virus genomes and absent from human DNA”
Bioinformatics, vol. 31, pp. 2421-2425, August 2015, pdf - E. D. Coelho, A. M. Santiago, J. P. Arrais, and J. L. Oliveira
“Computational methodology for predicting the landscape of the human-microbial interactome region level influence”
J Bioinform Comput Biol, August 2015, pdf - J. A. Vanegas, S. Matos, F. A. Gonzalez, J. L. Oliveira
“An Overview of Biomolecular Event Extraction from Scientific Documents”
Comp. and Mathematical Methods in Medicine, August 2015. pdf - D. Pratas, R. M. Silva, A. J. Pinho, and P. J. S. G. Ferreira
“An alignment-free method to find and visualise rearrangements between pairs of DNA sequences”
Scientific Reports, vol. 5, May 2015. pdf - R. M. Silva, D. Pratas, L. Castro, A. J. Pinho, P. J. Ferreira
“Three minimal sequences found in Ebola virus genomes and absent from human DNA”
Bioinformatics, April 2015. pdf - J. P. Arrais and J. L. Oliveira
“RecRWR: A Recursive Random Walk Method for Improved Identification of Diseases”
BioMed Research International, March 2015. pdf -
D. Campos, S. Matos, and J. L. Oliveira“A document processing pipeline for annotating chemical entities in scientific documents”Journal of Cheminformatics, 7(Suppl 1):S7, January 2015. pdf
- M. Krallinger, O. Rabal, F. Leitner, M. Vazquez, D. Salgado, Z. Lu, R. Leaman, Y. Lu, D. Ji, D. M. Lowe, R. A. Sayle, R. T. Batista-Navarro, R. Rak, T. Huber, T. Rocktäschel, S. Matos, D. Campos, B. Tang, H. Xu, T. Munkhdalai, K. H. Ryu, S. V.Ramanan, S. Nathan, S. Žitnik, M. Bajec, L. Weber, M. Irmer, S. A. Akhondi, J. A. Kors, S. Xu, X. An, U. K. Sikdar, A. Ekbal, M. Yoshioka, T. M. Dieb, M. Choi, K. Verspoor, M. Khabsa, C. L. Giles, H. Liu, K. E. Ravikumar, A. Lamurias, F. M. Couto, H. Dai, R. T.-H. Tsai, C. Ata, T. Can, A. Usié, R. Alves, I. Segura-Bedmar, P. Martínez, J. Oyarzabal, A. Valencia
“The CHEMDNER corpus of chemicals and drugs and its annotation principles”
Journal of Cheminformatics, vol. 7, Suppl. 1, p. S2, January 2015. pdf - L. Ragionieri, R. Vitorino, J. Frommlet, J. L. Oliveira, P. Gaspar, L. R. de Pouplana, et al
“Improving the accuracy of recombinant protein production through integration of bioinformatics, statistical and mass spectrometry methodologies”
FEBS Journal, December 2014. pdf - T. M. Godinho, C. Viana-Ferreira, L. B. Silva, and C. Costa
“A Routing Mechanism for Cloud Outsourcing of Medical Imaging Repositories”
IEEE Journal of Biomedical and Health Informatics, October 2014. pdf - S. Brás, A. Georgakis, L. Ribeiro, D. A. Ferreira, A. Silva, L. Antunes, and C. S. Nunes
“Electroencephalogram-based indices applied to dogs’ depth of anaesthesia monitoringe”
Research in Veterinary Science, October 2014. pdf - S. Duarte-Pereira, S. S. Silva, L. Azevedo, L. Castro, A. Amorim, and R. M. Silva
“Nampt and Naprt1: Novel Polymorphisms and Distribution of Variants between Normal Tissues and Tumor Samples”
Scientific Reports, Vol. 4,:6311, September 2014. pdf - R. Sebastião, J. Gama, and T. Mendonça
“Constructing fading histograms from data streams”
Progress in Artificial Intelligence, August 2014. pdf - L. B. Silva, S. Campos, C. Costa, and J. L. Oliveira
“Sensor-Based Architecture for Medical Imaging Workflow Analysis”
Journal of Medical Systems, August 2014. pdf - D. Campos, J. Lourenco, S. Matos, and J. L. Oliveira
“Egas: a collaborative and interactive document curation platform”
Database, June 2014. pdf - N. Rosa, M. J. Correia, J. P. Arrais, N. Costa, J. L. Oliveira, and M. Barros
“The Landscape of Protein Biomarkers Proposed for Periodontal Disease: Markers with Functional Meaning”
BioMed Research International, June 2014. pdf - E. Coelho, J. P. Arrais, S. Matos, C. Pereira, N. Rosa, M. J. Correia, M. Barros, and J. L. Oliveira
“Computational prediction of the human-microbial oral interactome”
BMC Systems Biology, February 2014. pdf - J. C. Santos and S. Matos
“Analysing Twitter and web queries for flu trend prediction”
Theoretical Biology and Medical Modelling, vol. 11 (Suppl 1):S6, May 2014, pdf - V. M. Prieto, S. Matos, M. Álvarez, F. Cacheda, and J. L. Oliveira
“Twitter: A Good Place to Detect Health Conditions”
PLoS ONE, January 2014. pdf - M. Reboiro-Jato, J. P. Arrais, J. L. Oliveira, and F. Fdez-Riverola
“geneCommittee: a web-based tool for extensively testing the discriminatory power of biologically relevant gene sets in microarray data classification”
BMC Bioinformatics, January 2014. pdf (highly accessed) - D. Campos, Q. C. Bui, S. Matos, and J. L. Oliveira
“TrigNER: automatically optimized biomedical event trigger recognition on scientific documents”
Source Code for Biology and Medicine, vol. 9:1, January 2014. pdf - P. Lopes, T. Nunes, D. Campos, L. Furlong, A. Bauer-Mehren, F. Sanz, M. C. Carrascosa, J. Mestres, J. Kors, B. Singh, E. van Mulligen, J. Van der Lei, G. Diallo, P. Avillach, E. Ahlberg, S. Boyer, C. Diaz, and J. L. Oliveira
” Gathering and Exploring Scientific Knowledge in Pharmacovigilance”
PLoS ONE, 2013. pdf - L. Ribeiro, C. Viana-Ferreira, J. L. Oliveira, and C. Costa
” XDS-I outsourcing proxy: ensuring confidentiality while preserving interoperability”
IEEE Journal of Biomedical and Health Informatics, 2013. pdf - P. Gaspar, P. Lopes, J. Oliveira, R. Santos, R. Dalgleish, and J. L. Oliveira
“Variobox: Automatic Detection and Annotation of Human Genetic Variants”
Human Mutation , 2013. pdf - L. B. Silva, R. Pinho, L. Ribeiro, C. Costa, and J. L. Oliveira
“A Centralized Platform for Geo-Distributed PACS Management”
Journal of Digital Imaging , 2013. pdf - S. Matos, H. Araújo, and J. L. Oliveira
“Biomedical literature exploration through latent semantics”
Advances in Distributed Computing and Artificial Intelligence Journal, Vol. 1(5):65-74, August 2013. pdf - L. A. Campos, V. L. Pereira Jr., A. Muralikrishna, S. Albarwani, S. Brás, and S. Gouveia
“Mathematical Biomarkers for the Autonomic Regulation of Cardiovascular System”
Frontiers in Integrative Physiology, 2013. pdf - D. Campos, S. Matos, and J. L. Oliveira
“A modular framework for biomedical concept recognition”
BMC Bioinformatics, 14:281, 2013. pdf (highly accessed) - P. M. Coloma, M. J. Schuemie, G. Trifirò, L. Furlong, E.van Mulligen, A. Bauer-Mehren, P. Avillach, J. Kors, F. Sanz, J. Mestres, J. L. Oliveira, S. Boyer, E. A. Helgee, M. Molokhia, J. Matthews, D. Prieto-Merino, R. Gini, R. Herings, G. Mazzaglia, G. Picelli, L. Scotti, L. Pedersen, J. van der Lei, M. Sturkenboom
“Drug-Induced Acute Myocardial Infarction: Identifying ‘Prime Suspects’ from Electronic Healthcare Records-Based Surveillance System”
PLoS ONE , 2013. pdf - P . Lopes. and J. L. Oliveira
“An innovative portal for rare genetic diseases research: The semantic Diseasecard”
Journal of Biomedical Informatics, 2013. pdf - M. Cases, L. I. Furlong, J. Albanell, R. B. Altman, R. Bellazzi, S. Boyer, A. Brand, A. J. Brookes, S. Brunak, T. W. Clark, J. Gea, P. Ghazal, N. Graf, R. Guigó, T. E. Klein, N. López-Bigas, V. Maojo, B. Mons, M. Musen, J. L. Oliveira, A. Rowe, P. Ruch, A. Shavo, E. H. Shortliffe, A. Valencia, J. v. d. Lei, M. A. Mayer, and F. Sanz
“How to improve data and knowledge management to better integrate healthcare and research”
Journal of Internal Medicine, 2013. pdf - T. Nunes, D. Campos, S. Matos, and J. L. Oliveira
“BeCAS: biomedical concept recognition services and visualization”
Bioinformatics, Vol. 29(15):1915-6, August 2013. pdf - F. Valente, C. Costa, and A. Silva
“Dicoogle, a PACS featuring Profiled Content Based Image Retrieval”
PLoS ONE, 2013. pdf - K. K. Lee, S. Matos, D. H. Evans, P. White, I. D. Pavord, S. S. Birring
“A Longitudinal Assessment of Acute Cough”
American Journal of Respiratory and Critical Care Medicine, vol. 187, no. 9, p. 991-997, May 2013. pdf - J. P. Arrais, N. Rosa, J. Melo, E. Coelho, D. Amaral, M. J. Correia, M. Barros, and J. L. Oliveira
“OralCard: a bioinformatic tool for the study of Oral Proteome”
Archives of Oral Biology, February 2013. pdf - D. Campos, S. Matos, and J. L. Oliveira
“Gimli: open source and high-performance biomedical name recognition”
BMC Bioinformatics, 14:54, February 2013. pdf (highly accessed) - P. Gaspar, G. Moura, M. A. S. Santos, and J. L. Oliveira
“mRNA secondary structure optimization using a correlated stem-loop prediction”
Nucleic Acids Research, January 2013. pdf - N. Yousaf, W. Monteiro, S. Matos, S. Birring, and I. D. Pavord
“Cough frequency in health and disease”
European Respiratory Journal, vol. 41, no. 1, p. 241-243, January 2013. pdf - P. Lopes and J. L. Oliveira
“COEUS: “semantic web in a box” for biomedical applications”
Journal of Biomedical Semantics, vol. 3, December 2012. pdf (highly accessed) - J. L. Oliveira, P. Lopes, T. Nunes, D. Campos, S. Boyer, E. Ahlberg, E. van Mullingen, J. Kors, B. Singh, L. I. Furlong, F. Sanz, A. Bauer-Mehren, M. D. C. Carrascosa, J. Mestres, P. Avillach, C. Díaz Acedo, and J. van der Lei
“The EU-ADR Web Platform: delivering advanced pharmacovigilance tools”
Pharmacoepidemiology and Drug Safety, December 2012. pdf - J. P. Lousado, J. L. Oliveira, G. Moura, and M. A. S. Santos
“An integrative approach for codon repeats evolutionary analyses”
International Journal of Data Mining and Bioinformatics, vol. 6: 369-381, September 2012. pdf - C. Costa and J. L. Oliveira
“Telecardiology through ubiquitous Internet services”
International Journal of Medical Informatics, vol. 81: 612-21, September 2012. pdf - L. B. Silva, C. Costa, and J. L. Oliveira
“DICOM Relay over the Cloud”
International Journal of Computer Assisted Radiology and Surgery, August 2012. pdf - P. Gaspar, J. L. Oliveira, J. Frommlet, M. A. S. Santos, and G. Moura
“EuGene: Maximizing synthetic gene design for heterologous expression”
Bioinformatics, vol. 28, July 2012. pdf - K. K. Lee, A. Savani, S. Matos, D. H. Evans, I. D. Pavord, S. Birring
“4-hour cough frequency monitoring in chronic cough”
Chest, July 2012. pdf - L. Ribeiro, C. Costa, and J. L. Oliveira
“Clustering of distinct PACS archives using a cooperative peer-to-peer network”
Computer Methods and Programs in Biomedicine, June 2012. pdf - F. Valente, C. Viana-Ferreira, C. Costa, and J. L. Oliveira
“A RESTful Image Gateway for Multiple Medical Image Repositories”
IEEE Transactions on Information Technology in BioMedicine, vol. 16, no. 3: 356-36, 2012. pdf - D. Campos, S. Matos, I. Lewin, J. L. Oliveira, and D. Rebholz-Schuhmann
“Harmonisation of gene/protein annotations: towards a gold standard MEDLINE”
Bioinformatics, Vol. 28(9):1253-61, May 2012. pdf - A. Bauer-Mehren, E. van Mullingen, P. Avillach, M. D. C. Carrascosa, R. García-Serna, J. Pinero, B. Singh, P. Lopes, J. L. Oliveira, G. Diallo, A. E. Helgee, B. Scott, J. Mestres, F. Sanz, J. Kors, and L. I. Furlong
“Automatic filtering and substantiation of drug safety signals”
PLoS Computational Biology, 2012. pdf - P. Gaspar and J. L. Oliveira
“Advantages of a Pareto-based genetic algorithm to solve the gene synthetic design problem”
Current Bioinformatics, vol. 7, no. 4, 2012. - S.C. Novais, T. Vandenbrouck, P. Lopes, J. P. Arrais, W. De Coen, A. Soares and M. Amorim
“Enchytraeus albidus microarray: enrichment, design, annotation and database (EnchyBASE)”
PLoS ONE, 2012. pdf - N. Rosa, M. J. Correia, J. P. Arrais, P. Lopes, J. Melo, J. L. Oliveira, and M. Barros
“From the salivary proteome to the OralOme: Comprehensive molecular oral biology”
Archives of Oral Biology, 2012. pdf - P. Grynberg, T. Abeel, P. Lopes, G. Macintyre, and L. P. Rubiño
“Highlights from the Student Council Symposium 2011 at the International Conference on Intelligent Systems for Molecular Biology and European Conference on Computational Biology”
BMC Bioinformatics, 12 (Suppl 11):A1, November 2011. pdf - G. Moura, M. Pinheiro, A. Freitas, J. L. Oliveira, J. Frommlet, L. Carreto, A. R. Soares, A. R. Bezerra, and M. A. S. Santos
“Species-Specific Codon Context Rules Unveil Non-Neutrality Effects of Synonymous Mutations”
PLoS ONE, vol. 6, 2011.
Conferences
- R. Lebre, M. Pedrosa, C. Costa
“A safe architecture for authorisation grant in healthcare ecosystems”
In ICHI 2020 – IEEE International Conference on Healthcare Informatics 2020, Oldenburg, Germany, December 2020. - A. Trifan, R. Antunes and J. L. Oliveira
“Machine learning for depression screening in online communities”
In 14th International Conference on Practical Applications of Computational Biology & Bioinformatics, PACBB 2020, L’Aquila, Italy, October 2020. - R. Lebre, E. Pinho, J. M. Silva, C. Costa
“Dicoogle Framework for Medical Imaging Teaching and Research”
In ICTS4eHealth 2020 – IEEE International Workshop on ICT Solutions for E-Health, Rennes, France, June 2020. - A. Trifan, R. Antunes, S. Matos and J.L. Oliveira
“Understanding depression from psycholinguistics patterns in social media texts”
In ECIR2020 – European Conference on Information Retrieval, Lisbon, Portugal, April 2020. - T. Almeida and S. Matos
“Calling Attention to Passages for Biomedical Question Answering”
In ECIR2020 – European Conference on Information Retrieval, Lisbon, Portugal, April 2020. - R. Jesus, P. Nunes, R. Lebre, C. Costa
“Role-based architecture for secure management of telepathology sessions”
In MIE2020 – Medical Informatics Europe, Geneva, Switzerland, April 2020. - A. Trifan, D. Semeraro, J. Drake, R. Bukowski and J.L. Oliveira
“Social media mining for post-partum depression prediction”
In MIE2020 – Medical Informatics Europe, Geneva, Switzerland, April 2020. - J. R. Almeida, E. Monteiro, L. B. Silva, A. Pazos, J. L. Oliveira
“A Recommender System based on Cohorts’ Similarity”
In The 30th Medical Informatics Europe conference, Geneva, Switzerland, April 2020. - J.F. Silva, R. Antunes, J.R. Almeida and S. Matos
“Clinical concept normalization on medical records using word embeddings and heuristics”
In The 30th Medical Informatics Europe conference, Geneva, Switzerland, April 2020. - J. R. Almeida and S. Matos
“Rule-based extraction of family history information from clinical notes”
In The 35th ACM/SIGAPP Symposium On Applied Computing, Brno, Czech Republic, March 2020. - R. Antunes, J.F. Silva and S. Matos
“Evaluating semantic textual similarity in clinical sentences using deep learning and sentence embeddings”
In The 35th ACM/SIGAPP Symposium On Applied Computing, Brno, Czech Republic, March 2020. - J. R. Almeida, P. Freire, O. Fajarda and J. L. Oliveira
“A Computational Platform for Heart Failure Cases Research”
In Healthinf 2020 – International Conference of Health Informatics, Valletta, Malta, February 2020. - J. R. Almeida, J. F. Silva, A. Sierra, S. Matos and J. L. Oliveira
“Enhancing Decision-making Systems with Relevant Patient Information by Leveraging Clinical Notes”
In Healthinf 2020 – International Conference of Health Informatics, Valletta, Malta, February 2020. - P. Nunes, R. Jesus, R. Lebre and C. Costa
“Data and Sessions Management in a Telepathology Platform”
In Healthinf 2020 – International Conference of Health Informatics, Valletta, Malta, February 2020. - A.J. Neves, R. Ribeiro and J. L. Oliveira
“Image Selection based on Low Level Properties for Lifelog Moment Retrieval”
In 12th International Conference on Machine Vision, ICMV 2019, Amsterdam, The Netherlands, November 2019. - M. Pedrosa, A. Zúquete and C. Costa
“Pseudonymisation with break-the-glass compatibility for health records in federated services”
In 19th IEEE International Conference on BioInformatics and BioEngineering (IEEE BIBE 2019), Athens, Greece, October 2019. - F.S. Silva and R. Lebre
“Automated pattern recognition of Cerebral Amyloid Angiopathy patients”
In RecPad2019, Porto, Portugal, October 2019. - J.R. Almeida, P. Freire, O. Fajarda and J. L. Oliveira
“A Weighted Rule-Based Model for File Forgery Detection: UA. PT Bioinformatics at ImageCLEF 2019”
In CLEF 2019 Conference and Labs of the Evaluation Forum, Lugano, Switzerland, September 2019. - A. Trifan and J. L. Oliveira
“BioInfo@ UAVR at eRisk 2019: delving into social media texts for the early detection of mental and food disorders”
In CLEF 2019 Conference and Labs of the Evaluation Forum, Lugano, Switzerland, September 2019. - D. Canedo, A. Trifan and A.J. Neves
“Focus estimation in academic environments using Computer Vision”
In 9th Iberian Conference on Pattern Recognition and Image Analysis (ibPRIA 2019), Madrid, Spain, July 2019. - J. R. Almeida and J. L. Oliveira
“GenericCDSS – A Generic Clinical Decision Support System”
In IEEE 32th International Symposium on Computer-Based Medical Systems (CBMS), Córdoba, Spain, June 2019. - R. Lebre, R. Jesus, P. Nunes and C. Costa
“Collaborative Framework for a Whole-Slide Image Viewer”
In IEEE 32th International Symposium on Computer-Based Medical Systems (CBMS), Córdoba, Spain, June 2019. - M. Pedrosa, C. Costa and J. Dorado
“GDPR impacts and opportunities for computer-aided diagnosis – Guidelines and legal perspectives”
In IEEE 32th International Symposium on Computer-Based Medical Systems (CBMS), Córdoba, Spain, June 2019. - E. Pinho, J. Figueira Silva and C. Costa
“Volumetric feature learning for query-by-example in medical imaging archives”
In IEEE 32th International Symposium on Computer-Based Medical Systems (CBMS), Córdoba, Spain, June 2019. - R. Lebre, L.B. Silva and C. Costa
“An Accounting Mechanism for Standard Medical Imaging Services”
In 2019 IEEE 6th Portuguese Meeting on Bioengineering (ENBENG), Lisbon, Portugal, February 2019. - A. Trifan and J. L. Oliveira
“FAIRness in Biomedical Data Discovery”
In Proceedings of the 12th International Joint Conference on Biomedical Engineering Systems and Technologies, Prague, Czech Republic, February 2019. - J. R. Almeida, O. Fajarda, A. Pereira and J. L. Oliveira
“Strategies to Access Patient Clinical Data from Distributed Databases”
In Proceedings of the 12th International Joint Conference on Biomedical Engineering Systems and Technologies, Prague, Czech Republic, February 2019. - R. Antunes, J. F. Silva, A. Pereira and S. Matos
“Rule-based and machine learning hybrid system for patient cohort selection”
In Proceedings of the 12th International Joint Conference on Biomedical Engineering Systems and Technologies, Prague, Czech Republic, February 2019. - A. Teixeira, D. Pratas, A. J. Pinho and R. M. Silva
“Evolutionary insights from the comparative analysis of hominid genomes”
In Proceedings of the 24th Portuguese Conference on Pattern Recognition (RecPad2018), Coimbra, Portugal, October 2018. - C. Figueiredo, D. Pratas, A. J. Pinho, R. M. Silva
“Identification of antifungal targets using alignment-free methods”
In Proceedings of the 24th Portuguese Conference on Pattern Recognition (RecPad2018), Coimbra, Portugal, October 2018. - E. Pinho and C. Costa
“Feature Learning with Adversarial Networks for Concept Detection in Medical Images: UA.PT Bioinformatics at ImageCLEF 2018”
In Working notes of CLEF 2018 – Conference and Labs of the Evaluation Forum, Avignon, France, September 2018. - A.Trifan and J. L. Oliveira
“A FAIR marketplace for biomedical data custodians and clinical researchers”
In IEEE 31th International Symposium on Computer-Based Medical Systems (CBMS), Karlstad, Sweden, June 2018. - J. R. Almeida, J. Guimarães, and J. L. Oliveira
“Simplifying the digitization of clinical protocols for diabetes management”
In IEEE 31th International Symposium on Computer-Based Medical Systems (CBMS), Karlstad, Sweden, June 2018. - J. R. Almeida, T. M. Godinho, L. Bastião Silva, C. Costa, and J. L. Oliveira
“Services orchestration and workflow management in distributed medical imaging environments”
In IEEE 31th International Symposium on Computer-Based Medical Systems (CBMS), Karlstad, Sweden, June 2018. - J. M. Silva, A. Guerra, J. F. Silva, E. Pinho, and C. Costa
“Face De-Identification Service for Neuroimaging Volumes”
In IEEE 31th International Symposium on Computer-Based Medical Systems (CBMS), Karlstad, Sweden, June 2018. - J. F. Silva, J. M. Silva, A. Guerra, S. Matos, and C. Costa
“Ejection Fraction Classification in Transthoracic Echocardiography Using a Deep Learning Approach”
In IEEE 31th International Symposium on Computer-Based Medical Systems (CBMS), Karlstad, Sweden, June 2018. - C. Costa, M. Pedrosa, J. M. Silva and S. Matos
“Research PACS for diabetic retinopathy screening”
In Computer Assisted Radiology and Surgery Proceedings of the 32nd International Congress and Exhibition (CARS 2018), Berlin, Germany, June 2018. - J. M. Silva, A. Guerra, J. F. Silva, C. Costa and S. Matos
“A novel 3D-CNN approach for ejection fraction classification in transthoracic echocardiography”
In Computer Assisted Radiology and Surgery Proceedings of the 32nd International Congress and Exhibition (CARS 2018), Berlin, Germany, June 2018. - R. Lebre, T. Godinho, L. Silva and C. Costa
“A performant and fully DICOM compliant Web PACS for Digital Pathology”
In Computer Assisted Radiology and Surgery Proceedings of the 32nd International Congress and Exhibition (CARS 2018), Berlin, Germany, June 2018. - M. Pedrosa, J. M. Silva, S. Matos and C. Costa
“SCREEN-DR – Software architecture for the Diabetic Retinopathy Screening”
In Proceedings of MIE 2018, Medical Informatics Europe, Gothenburg, Sweden, April 2018. - R. Lebre, L. B. Silva and C. Costa
“Shared Medical Imaging Repositories”
In Proceedings of MIE 2018, Medical Informatics Europe, Gothenburg, Sweden, April 2018. - F. Maia, L. B. Silva and J. L. Oliveira
“Biomedical Informatics – How to choose the best tool for each task”
In Proceedings of MIE 2018, Medical Informatics Europe, Gothenburg, Sweden, April 2018. - A. Trifan, J. van der Lei, C. Diaz and J. L. Oliveira
“A Methodology for fine-grained access control in Exposing Biomedical Data”
In Proceedings of MIE 2018, Medical Informatics Europe, Gothenburg, Sweden, April 2018. - M. Pedrosa, J. Miguel and C. Costa
“Reactive Through Services – Opinionated Framework for Developing Reactive Services”
In Proceedings of the 8th International Conference on Cloud Computing and Services Science – Volume 1: CLOSER, Funchal, Madeira, Portugal, March 2018. - J. R. Almeida, R. Ribeiro, and J. L. Oliveira
“A modular workflow management framework”
In Proceedings of the 11th International Conference on Health Informatics (HealthInf 2018), Funchal, Madeira, Portuga, January 2018. - O. Fajarda; L. B. Silva, P. Rijnbeek, M. Van Speybroeck, and J. L. Oliveira
“A methodology to perform semi-automatic distributed EHR database queries.”
In Proceedings of the 11th International Conference on Health Informatics (HealthInf 2018), Funchal, Madeira, Portuga, January 2018. - J. M. Silva, A. Silva and P. Vilela
“Enhanced cerebral vascular segmentation with harmonic constraints”
In 23rd Portuguese Conference on Pattern Recognition, Lisbon, Portugal, October, 2017. - R. Antunes and S. Matos
“Evaluation of word embedding vector averaging functions for biomedical word sense disambiguation”
INForum 2017 – Atas do Nono Simpósio de Informática, Aveiro, p. 25-30, October 2017. - S. Matos and R. Antunes
“Identifying Relevant Literature for Precision Medicine Using Deep Neural Networks”
Proceedings of the VI BioCreative Challenge Evaluation Workshop, Bethesda, MA, USA, p. 99-101, October 2017 - R. Antunes and S. Matos
“Biomedical Word Sense Disambiguation with Word Embeddings”
11th International Conference on Practical Applications of Computational Biology & Bioinformatics, Porto, p. 273-279, June 2017 - J.F. Silva, J.M. Silva, E. Pinho and C. Costa
“3D-CNN in drug resistance detection and tuberculosis classification”
In CEUR Workshop Proceedings (Vol. 1866), 2017 - E. Pinho, J. F. Silva, J. M. Silva, C. Costa
“Towards representation learning for biomedical concept detection in medical images: UA.PT bioinformatics in ImageCLEF ”
In CEUR Workshop Proceedings (Vol. 1866), 2017 - P. Sernadela and J. L. Oliveira
“Automated nanopublications generation from biomedical literature”
In IEEE 5th Portuguese Meeting on Bioengineering (ENBENG), Coimbra, Portugal, February 2017 - E. Pinho and C. Costa
“Extensible Architecture for Multimodal Information Retrieval in Medical Imaging Archives”
In International Conference on Signal Image Technology and Internet-based Systems (SITIS), Naples, Italy, November 2016 - A. Santos, S. Matos, D. Campos and J. L. Oliveira
“A Curation Pipeline and Web-Services for PDF Documents”
In Proceedings of the 7th International Symposium on Semantic Mining in Biomedicine (SMBM 2016), Potsdam, Germany, August 2016 - V. Afreixo, J.M.O.S. Rodrigues, C.A.C. Bastos, R.M. Silva
“Exceptional Symmetry Profile: A Genomic Word Analysis”
In International Conference on Practical Applications of Computational Biology & Bioinformatics (PACBB), Sevilla, Spain, June 2016 - E. Monteiro, P. Sernadela, S. Matos, C. Costa and J. L. Oliveira
“Semantic Knowledge Base Construction from Radiology Reports”
In Proceedings of the 9th International Joint Conference on Biomedical Engineering Systems and Technologies, Rome, Italy, February 2016 - P. Sernadela, S. Matos, J. L. Oliveira
“Ann2RDF: moving annotations to semantic web”
In 17th International Conference on Information Integration and Web-based Applications & Services (iiWAS 2015), Brussels, Belgium, December 2015 - P. Lopes, L. Bastião and J. L. Oliveira
“i2x: an Automated Real-time Integration and Interoperability Platform”
In Proceedings of the 8th IEEE International Conference on Service Oriented Computing & Applications (SOCA2015), Rome, Italy, October 2015 - D. Pratas, RM. Silva, AJ. Pinho, PJSG. Ferreira
“Detection and visualisation of regions of human DNA not present in other primates”
In Proceedings of the 21st Portuguese Conference on Pattern Recognition, RecPad 2015, Faro, Portugal, October 2015 - S. Matos, D. Campos, R. Pinho, R. Silva, M. Mort, D. Cooper, J. L. Oliveira
“Assisted Mining and Curation of Genomic Variants using Egas”
In Proceedings of the Fifth BioCreative Challenge Evaluation Workshop, Sevilla, Spain, September 2015 - S. Matos, J. Sequeira, D. Campos, J. L. Oliveira
“Identification of chemical and gene mentions in patent texts using feature-rich conditional random fields”
In Proceedings of the Fifth BioCreative Challenge Evaluation Workshop, Sevilla, Spain, September 2015 - S. Matos, A. Santos, D. Campos, J. L. Oliveira
“Neji: a BioC compatible framework for biomedical concept recognition”
In Proceedings of the Fifth BioCreative Challenge Evaluation Workshop, Sevilla, Spain, September 2015 - L. A. Bastiao Silva, C. Días, J. van der Lei, and J. L. Oliveira
“Architecture to Summarize Patient-Level Data Across Borders and Countries”
In Proceedings of MEDINFO 2015, Brasil, August 2015 - E. Coelho, J. P. Arrais, and J. L. Oliveira
“Uncovering Microbial Duality within Human Microbiomes: A Novel Algorithm for the Analysis of Host-Pathogen Interactions”
In Proceedings of the 37th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC2015), Milan, Italy, August 2015 - J. Arevalo, F. A. Gonzalez Osorio, R. Ramos-Pollan, J. L. Oliveira, and M. A. Guevara López
“Convolutional Neural Networks for Mammography Mass Lesion Classification”
In Proceedings of the 37th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC2015), Milan, Italy, August 2015 - E. Monteiro, C. Costa, J. L. Oliveira
“A Machine Learning Methodology for Medical Imaging Anonymization”
In Proceedings of the 37th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC2015), Milan, Italy, August 2015 - E. Pinho, C. V. Ferreira, and Carlos Costa
“Simulation of DICOM traffic in PACS networks using behavior profiles”
In Proceedings of Computer Assisted Radiology and Surgery (CARS 2015), Barcelona, Spain, June 2015 - S. Matos, J. Sequeira, and J.L. Oliveira
“BioinformaticsUA: Machine Learning and Rule-Based Recognition of Disorders and Clinical Attributes from Patient Notes”
In Proceedings of the 9th International Workshop on Semantic Evaluation (SemEval 2015), Denver, Colorado, USA, June 2015 - T. T. Godinho, L. M. Silva, and C. Costa
“An automation framework for PACS workflows optimization in shared environments”
In Proceedings of the 10th Iberian Conference onInformation Systems and Technologies (CISTI 2015), Aveiro, Portugal, June 2015 - P. Lopes and J. L. Oliveira
“Integration-as-a-service for bioinformatics”
In Proceedings of the 10th Iberian Conference on Information Systems and Technologies (CISTI 2015), Aveiro, Portugal, June 2015 - P. Lopes, P. Sernadela, and J. L. Oliveira
“Towards a knowledge federation of linked patient registries”
In Proceedings of the 10th Iberian Conference on Information Systems and Technologies (CISTI 2015), Aveiro, Portugal, June 2015 - P. Lopes, L. Bastiao, and J. L. Oliveira,
“Challenges and opportunities for exploring patient-level data: Preliminary results”
In Proceedings of the 10th Iberian Conference on Information Systems and Technologies (CISTI 2015), Aveiro, Portugal, June 2015 - P. Sernadela, A. Pereira, and R. Rossetti
“DISim: Ontology-driven simulation of biomedical data integration tasks”
In Proceedings of the 10th Iberian Conference on Information Systems and Technologies (CISTI 2015), Aveiro, Portugal, June 2015 - C. Viana-Ferreira, S. Matos, C. Costa
” Long-Term Prefetching for Cloud Medical Imaging Repositories”
In Proceedings of the 26th Medical Informatics Europe Conference (MIE2015), Madrid, Spain, May 2015 - E. Monteiro, F. Valente, J. L. Oliveira, C. Costa
“A Recommender System for Medical Imaging Diagnostic”
In Proceedings of the 26th Medical Informatics Europe Conference (MIE2015), Madrid, Spain, May 2015 - P. Sernadela, P. Lopes, D. Campos, S. Matos, J. L. Oliveira
“A Semantic Layer for Unifying and Exploring Biomedical Document Curation Results”
In Third International Conference (IWBBIO 2015), Bioinformatics and Biomedical, Granada, Spain, April 2015 - S. Brás, A. Silva, J. Ribeiro, and J. Oliveira
“New Insights in Echocardiography Based Left-Ventricle Dynamics Assessment”
In Third International Conference (IWBBIO 2015), Bioinformatics and Biomedical, Granada, Spain, April 2015 - P. Lopes and J. Oliveira
“An Event-Driven Architecture for Biomedical Data Integration and Interoperability”
In Third International Conference (IWBBIO 2015), Bioinformatics and Biomedical Engineering, Granada, Spain, April 2015 - C. V. Ferreira, S. Matos, and Carlos Costa
“Incremental Learning Versus Batch Learning for Classification of User’s Behaviour in Medical Imaging”
In Proceedings of the 8th International Conference on health informatics (HealthInf 2015), Lisbon, Portugal, January 2015 - L. Bastião, C. Costa, and J. L. Oliveira
“Semantic search over DICOM repositories”
In IEEE International Conference on Healthcare Informatics 2014 (ICHI 2014) Verona, Italy. September 2014 - F. Valente, A. Silva, C. Costa, J. M. F. Valiente, C. Suárez-Ortega, M. Guevara
“A dataflow-based appraoch to the design and distribution of medical image analytics”
In Proceedings of 8th Iberian Grid Infrastructure Conference (IBERGRID 2014)”, p. 201-212, Aveiro, Portugal, September 2014 - S. Matos, T. Nunes, and J.L. Oliveira
“BioinformaticsUA: Concept Recognition in Clinical Narratives Using a Modular and Highly Efficient Text Processing Framework”
In Proceedings of the 8th International Workshop on Semantic Evaluation (SemEval 2014), p. 135–139, Dublin, Ireland, August 2014 - T. Godinho, L. Bastião, C. Costa, and J. L. Oliveira
“Multi-provider Architecture for Cloud Outsourcing of Medical Imaging Repositories”
In 25th European Medical Informatics Conference (MIE2014) Istanbul, Turkey, August 2014 - L. Bastião, M. Santos, L. Ribeiro, C. Costa, and J. L. Oliveira
“Screening Radiation Exposure for Quality Assurance”
In 25th European Medical Informatics Conference (MIE2014). Istanbul, Turkey, August 2014 - E. Pinho, L. Bastiao Silva, and C. Costa
“A cloud service integration platform for web applications”
In Proccedings of the International Conference on High Performance Computing & Simulation (HPCS 2014), Bologna, Italy, July 2014 - C. V. Ferreira, and C. Costa
“DICOM traffic generator based on behaviour profiles”
In IEEE International Conference on Biomedical and Health Informatics (BHI 2014), p. 93-96, Valencia, Spain, June 2014 - T. Nunes, S. Matos, and J.L. Oliveira
“Extracting sentences describing biomolecular events from the biomedical literature”
In Proceedings of the 11th International Symposium on Distributed Computing and Artificial Intelligence (DCAI 2014), Salamanca, Spain, June 2014 - L. Bastião, L. Bernoud, C. Costa, and J. L. Oliveira
“NoSQL medical imaging archive: comparison between different implementations”
In IEEE International Conference on Biomedical and Health Informatics (BHI 2014) Valencia, Spain, June 2014 - L. Bastião, M. Santos, C. Costa, and J. L. Oliveira
“Normalizing medical imaging archives for dose quality assurance and productivity auditing”
In IEEE International Symposium on Medical Measurement and Applications (MeMeA 2014) Lisbon, Portugal, June 2014 - S. Brás, L. Ribeiro, D. A. Ferreira, L. Antunes, and C. Nunes
“Controlling the Hypnotic Drug (propofol) to maintain a stable depth of Anesthesia, in Dogs”
In Proccedings of IEEE International Symposium on Medical Measurements and Applications (MeMeA 2014), Lisbon, Portugal, June 2014 - R.Correia, S. Brás, A. Silva, J. Mendes, D. A. Ferreira
“Development of new research software for real-time raw electroencephalogram analysis”
In Proccedings of IEEE International Symposium on Medical Measurements and Applications (MeMeA 2014), Lisbon, Portugal, June 2014 - L. Castro, J. L. Oliveira, and R. M. Silva
“Mutation Analysis in Park2 Gene Uncovers Patterns of Associated Genetic Variants”, Advances in Intelligent Systems and Computing,
In Proceedings of the 8th Internation Conference on Practical Applications of Computational Biology & Bioinformatics (PACBB 2014), Salamanca, Spain, June 2014 - P. Sernadela, E. van der Horst, M. Thompson, P. Lopes, M. Roos, and J. L. Oliveira
“A Nanopublishing Architecture for Biomedical Data”
In Proceedings of the 8th International Conference on Practical Applications of Computational Biology & Bioinformatics(PACBB 2014), Salamanca, Spain, June 2014 - F. Barbosa, J. Arrais and J.L. Oliveira
“Weighted Gene Co-expression Network Analysis Applied to Head and Neck Squamous Cell Carcinoma Data”
In Proceedings of the International Conference on Health Informatics (ICHI 2013), Vilamoura, Portugal, November 2013 - E. Monteiro, C. Costa, and J. L. Oliveira
“A DICOM viewer based on web technology”
In Proceedings of IEEE 15th International Conference on e-Health Networking, Applications & Services (Healthcom 2013), Lisboa, Portugal, October 2013 - C. V. Ferreira, and C. Costa
“A cloud based architecture for medical imaging services”
In Proceedings of IEEE 15th International Conference on e-Health Networking, Applications & Services (Healthcom 2013), Lisboa, Portugal, October 2013 - L. Ribeiro, F. Honório, J. L. Oliveira, C. Costa
“Leveraging XDS-I and PIX workflows for validating cross-enterprise patient identity linkage”
In Proceedings of IEEE 15th International Conference on e-Health Networking, Applications & Services (Healthcom 2013), Lisboa, Portugal, October 2013 - D. Campos, S. Matos, and J. L. Oliveira
“Chemical name recognition with harmonized feature-rich conditional random fields”
In Proceedings of Fourth BioCreative Challenge Evaluation Workshop, vol. 2, p. 82-87, October 2013 - D. Campos, J. Lourenço, T. Nunes, R. Vitorino, P. Domingues, S. Matos, and J. L. Oliveira
“Egas – Collaborative Biomedical Annotation as a Service”
In Proceedings of Fourth BioCreative Challenge Evaluation Workshop, vol. 1, p. 254-259, October 2013 - L. Bastião, C. Costa, and J. L. Oliveira
“An agile framework to support distributed medical imaging scenarios”
In IEEE International Conference on Healthcare Informatics 2013 (ICHI 2013) Philadelphia, PA, USA. September 2013 - F. Marques, P. Azevedo, J. P. Cunha, M. B. Cunha, S. Brás, and J. M. Fernandes
“IREMAN: FIRefighter team brEathing Management system using ANdroid”
In 17th International Symposium on Wearable Computers (ISWC 2013), Zurich, Switzerland, September 2013 - L. Ribeiro, R. Rodrigues, C. Costa and J.L. Oliveira,
“Enabling Outsourcing XDS for Imaging on the Public Cloud”
In Proceedings of the 14th World Congress on Medical and Health Informatics (MEDINFO 2013), Copenhagen, Denmark, August 2013 - D. Campos, S. Matos, and J. L. Oliveira
“Neji: a tool for heterogeneous biomedical concept identification”
In Proceedings of the BioLINK SIG, ISMB/ECCB, Berlin, Germany, July 2013 - L. Bastiao, S. Campos, C. Costa, and J. L.Oliveira
“Integrating echocardiogram reports with medical imaging”
In 26th IEEE International Symposium on Computer-Based Medical Systems (CBMS 2013), Porto, Portugal, June 2013 - S. Brás, J. M. Fernandes, and J. P.S. Cunha,
“ECG Delineation and Morphological Analysis for Firefighters Tasks Differentiation”
In Proceedings of the 26th International Symposium on Computer-Based Medical Systems (CBMS 2013), Porto, Portugal, June 2013 - M. Santos, L. Bastião, C. Costa, A. Silva, and N. Rocha,
“Multi Vendor DICOM Metadata Access: A Multi Site Hospital Approach Using Dicoogle”
In 8th Iberian Conference on Information Systems and Technologies (CISTI 2013), Lisboa, Portugal, June 2013 - T. Godinho, L. B. Silva, C. Viana-Ferreira, C. Costa and J. L. Oliveira
“Enhanced regional network for medical imaging repositories”
In 8th Iberian Conference on Information Systems and Technologies (CISTI 2013), Lisboa, Portugal, June 2013 - V M. Prieto, S. Matos, M. Álvarez, F.Cacheda, and J. L. Oliveira
“Analysing Relevant Diseases From Iberian Tweets”
In Proceedings of the 7th Int. Conf. on Practical Applications of Computational Biology and Bioinformatics (PACBB 2013), Salamanca, Spain, May 2013 - S. Matos, H. Araújo, and J. L. Oliveira
“Structuring and Exploring the Biomedical Literature Using Latent Semantics”
In Proceedings of the 10th International Symposium on Distributed Computing and Artificial Intelligence (DCAI 2013), Salamanca, Spain, May 2013 - J. C. Santos and S. Matos
“Predicting Flu Incidence from Portuguese Tweets”
In Proceedings of the International Work-Conference on Bioinformatics and Biomedical Engineering, Granada, Spain, March 2013 - J. C. Santos, T. Pedrosa, C. Costa, and J. L. Oliveira
“Concepts for a Personal Health Record”
In 24th European Medical Informatics Conference (MIE 2012), Pisa, Italy, 2012. - C. Viana-Ferreira, D. Ferreira, F. Valente, E. Monteiro, C. Costa, and J. L. Oliveira
“Dicoogle Mobile: a medical imaging platform for Android”
In 24th European Medical Informatics Conference (MIE 2012), Pisa, Italy, 2012 - L. Ribeiro, C. Costa, and J. L. Oliveira
“Enhancing the many-to-many relations across IHE Document Sharing Communities”
In 24th European Medical Informatics Conference (MIE 2012), Pisa, Italy, 2012 - E. J. M. Monteiro, L. A. B. Silva, and C. Costa
“CloudMed: Promoting telemedicine processes over the cloud”
In Proceedings of the 7th Iberian Conference onInformation Systems and Technologies (CISTI 2012) 2012 - L. Bastião Silva, C. Costa, and J. L. Oliveira
“A DICOM relay service supported on cloud resources”
In 5th International Conference on Health Informatics (HEALTHINF 2012), Vilamoura, Portugal, 2012 - T. Pedrosa, R. P. Lopes, J. C. Santos, C. Costa, and J. L. Oliveira
“A secury personal health record repository”
In 5th International Conference on Health Informatics (HEALTHINF 2012), Vilamoura, Portugal, 2012 - L. Velte, T. Pedrosa, C. Costa, and J. L. Oliveira
“An OPENEHR repository based on a native XML database”
In 5th International Conference on Health Informatics (HEALTHINF 2012), Vilamoura, Portugal, 2012 - R. Mendonça, P. Lopes, H. Rocha, J. Oliveira, L. Vilarinho, R. Santos, and J. L. Oliveira
“Gathering and managing genotype and phenotype information about rare diseases patients”
In 5th International Conference on Health Informatics (HEALTHINF 2012), Vilamoura, Portugal, 2012 - J. Melo, J.P. Arrais, P. Lopes, N. Rosa, M. J. Correia, M. Barros, and J. L. Oliveira
“OralCard – web information system for oral health”
In 5th International Conference on Health Informatics (HEALTHINF 2012), Vilamoura, Portugal, 2012 - C. Viana-Ferreira, C. Costa, and J. L. Oliveira
“Dicoogle Relay – a Cloud Communications Bridge for Medical Imaging”
In 25th IEEE International Symposium on Computer-Based Medical Systems (CBMS 2012), Rome, Italy, 2012 - R. Mendonça, A. F. Rosa, J. L. Oliveira, and A. Teixeira
“Towards ontology based health information search in Portuguese – A case study in neurologic diseases”
In 7th Iberian Conference on Information Systems and Technologies (CISTI 2012), Madrid, Spain, 2012 - P. Gaspar, J. Carbonell, and J. L. Oliveira
“Parameter influence in genetic algorithm optimization of support vector machines”
In 7th International Conference on Practical Applications of Computational Biology & Bioinformatics (PACBB 2012), Salamanca, Spain, 2012 - P. Lopes, R. Mendonça, H. Rocha, J. Oliveira, L. Vilarinho, R. Santos, and J. L. Oliveira
“A Rare Disease Patient Manager”
In 7th International Conference on Practical Applications of Computational Biology & Bioinformatics (PACBB 2012), Salamanca, Spain, 2012 - P. Lopes and J. L. Oliveira
“COEUS: A Semantic Web Application Framework”
In Semantic Web Applications and Tools for Life Sciences (SWAT4LS 2011), London, UK, 2011 - S. Matos and J. L. Oliveira
“Prioritizing Literature Search Results Using a Training Set of Classified Documents”
In 5th International Conference on Practical Applications of Computational Biology & Bioinformatics (PACBB 2011), vol. 93, pp. 381-388, 2011. - P. Lopes and J. L. Oliveira
“Towards Knowledge Federation in Biomedical Applications”
In Proceedings of the 7th International Conference on Semantic Systems (I-Semantics 2011), Graz, Austria, 2011 - P. Lopes and J. L. Oliveira
“A Semantic Web Application Framework for Health Systems Interoperability”
In Proceedings of the first international workshop on Managing interoperability and complexity in health systems (MIXHS 2011), Graz, Austria, 2011 - C. Santos, T. Pedros, C. Costa, and J. L. Oliveira
“On the Use of Openehr in a Portable Phr”, Healthinf 2011: Proceedings of the International Conference on Health Informatics (Healthinf 2011), pp. 351-356, 2011. - T. Pedrosa, R. P. Lopes, J. C. Santos, C. Costa, and J. L. Oliveira
“Hybrid Electronic Health Records”, Healthinf 2011: Proceedings of the International Conference on Health Informatics (Healthinf 2011), pp. 571-574, 2011. - D. Campos, D. Rebholz-Schuhmann, S. Matos, and J. L. Oliveira
“A CRF-based approach to harmonize heterogeneous gene/protein annotations”
In Second CALBC Workshop, Hinxton, UK, 2011. - D. Campos, S. Matos, and J. L. Oliveira
“Annotating the CALBC corpus with a machine learning harmonization approach”
In Second CALBC Workshop, Hinxton, UK, 2011. - L. Bastião, C. Costa, A. Silva, and J. L. Oliveira
“A PACS Gateway to the Cloud”
In 6th Iberian Conference on Information Systems and Technologies (CISTI 2011), Chaves, Portugal, 2011, pp. 519-524. - J. Arrais and J. L. Oliveira
“Gene-disease prioritization through biomedical networks,” in 10th IEEE International Conference on Information Technology and Applications in Biomedicine (ITAB 2010), Corfu, Greece, 2010. - P. Lopes, D. Campos, and J. L. Oliveira
“A tagging system for bioinformatics resources,” in 10th IEEE International Conference on Information Technology and Applications in Biomedicine (ITAB 2010), Corfu, Greece, 2010. - J. Arrais and J. L. Oliveira
“On the exploitation of cloud computing in Bioinformatics,” in 10th IEEE International Conference on Information Technology and Applications in Biomedicine (ITAB 2010), Corfu, Greece, 2010. - P. Lopes and J. L. Oliveira
“An extensible platform for variome data integration,” in 10th IEEE International Conference on Information Technology and Applications in Biomedicine (ITAB 2010), Corfu, Greece, 2010. - L. S. Ribeiro, C. Costa, and J. L. Oliveira
“A Distributed and Reliable DICOM Storage Facility”
In 28th International EuroPACS Meeting (CARS 2010), Geneva, Switzerland, 2010. - F. Martin-Sanchez, V. Lopez-Alonso, L. Salamanca, J. L. Oliveira, and E. Andres
“Managing Knowledge Related to the Clinical Relevance of Biomarkers: An Example in Parkinson’s Disease”
In 2010 AMIA Summit on Translational Bioinformatics, San Francisco, CA, USA, 2010. - S. Matos, D. Campos, and J. L. Oliveira
“Vector-space models and terminologies in gene normalization and document classification”
In Proceedings of BioCreative III Workshop, Bethesda, Maryland, USA, September 2010 - S. Matos, J. Arrais, and J. L. Oliveira
“Expanding Gene-based PubMed Queries”
In 4th International Workshop on Practical Applications of Computational Biology and Bioinformatics (IWPACBB 2010), Guimarães, Portugal, 2010. - J. Arrais, J. Pereira, P. Lopes, S. Matos, and J. L. Oliveira
“Improving cross mapping in biomedical databases”
In 4th International Workshop on Practical Applications of Computational Biology and Bioinformatics (IWPACBB 2010), Guimarães, Portugal, 2010. - J. P. Lousado, J. L. Oliveira, G. Moura, and M. A. S. Santos
“An application for study tandem repeats in ortologous genes”
In 4th International Workshop on Practical Applications of Computational Biology and Bioinformatics (IWPACBB 2010), Guimarães, Portugal, 2010. - P. Lopes and J. L. Oliveira
“A Holistic Approach for Integrating Genomic Variation Information”
In Xth Spanish Symposium on Bioinformatics (JBI2010), Malaga, Spain, 2010. - J. P. Lousado, J. L. Oliveira, G. Moura, and M. A. S. Santos
“Análise da evolução de repetições de codões e de aminoácidos em dados biológicos”
In 5th Iberian Conference on Information Systems and Technologies (CISTI 2010), Santiago de Compostela, Spain, 2010. - D. Campos, S. Matos, and J. L. Oliveira
“Recognition of gene/protein names using Conditional Random Fields”
In International Joint Conference on Knowledge Discovery and Information Retrieval (KDIR 2010), Valencia, Spain, 2010. - L. S. Ribeiro, L. Bastião, C. Costa, and J. L. Oliveira
“Email-P2P Gateway to Distributed Medical Imaging Repositories”
In HEALTHINF 2010, Valencia, Spain, 2010. - J. C. Santos, T. Pedrosa, C. Costa, and J. L. Oliveira
“Modelling a Portable Personal Health Record”
In HEALTHINF 2010, Valencia, Spain, 2010. - T. Pedrosa, R. P. Lopes, J. C. Santos, C. Costa, and J. L. Oliveira
“Towards an EHR architecture for mobilde citizens”
In HEALTHINF 2010, Valencia, Spain, 2010. - J. C. Santos, T. Pedrosa, C. Ferreira, C. Costa, and J. L. Oliveira
“Gathering and Managing Complementary Diagnostic Tests,” in 5th Iberian Conference on Information Systems and Technologies (CISTI 2010), Santiago de Compostela, Spain, 2010. - L. S. Ribeiro, C. Costa, and J. L. Oliveira
“A Proxy of DICOM services”
In SPIE Medical Imaging 2010, S. Diego, CA, USA, 2010. - J. Arrais, J. Pereira, J. Fernandes, and J. L. Oliveira
“GeNS: A Biological Data Integration Platform”
In World Academy of Science, Engineering and Technology (WASET 2009), Venice, Italy, 2009, pp. 416 – 421. - P. Lopes and J. L. Oliveira
“Cloud Computing and Digital Libraries : First Perspectives on a Future Technological Alliance ”
In 9ª Conferência da Associação Portuguesa de Sistemas de Informação (CAPSI 2009), Viseu, Portugal, 2009. -
M. Pinheiro, M. J. Simões, C. Egas, and J. L. Oliveira
“Identifying SNPs candidates using 454 sequencing technology”
In Jornadas de Bioinformática (JB 2009), Lisbon, Portugal, 2009. - P. Lopes, J. Arrais, and J. L. Oliveira
“Link Integrator: A Link-based Data Integration Architecture”
In International Conference on Knowledge Discovery and Information Retrieval (KDIR 2009), Madeira, Portugal, 2009. - P. Lopes, D. Pinto, D. Campos, and J. L. Oliveira
“Arabella: A Directed Web Crawler”
In International Conference on Knowledge Discovery and Information Retrieval (KDIR 2009), Madeira, Portugal, 2009. - T. Pedrosa, C. Costa, R. P. Lopes, and J. L. Oliveira
“Virtual Health Card System”
In Inforum 2009, Lisbon, Portugal, 2009. - D. Polónia, C. Costa, J. L. Oliveira, and A. M. O. Duarte
“Inequality problems in the distribution of radiologists in Portugal: Requirements for the creation of an imaging marketplace”
In eChallenges 2009, Ankara, Turkey, 2009. - P. Lopes, J. Arrais, and J. L. Oliveira
“A Client-side Workflow Management System”
In 3rd International Workshop on Practical Applications of Computational Biology and Bioinformatics(IWPACBB 2009), Salamanca, Spain, 2009. - J. P. Lousado, G. Moura, M. A. S. Santos, and J. L. Oliveira
“Analysing the evolution of repetitive strands in genomes”
In 3rd International Workshop on Practical Applications of Computational Biology and Bioinformatics (IWPACBB 2009), Salamanca, Spain, 2009. - S. Matos, A. Barreiro, J.L. Oliveira
“Syntactic Parsing for Bio-Molecular Event Detection from Scientific Literature”, 14th Portuguese Conference on Artificial Intelligence, EPIA 2009, Aveiro, Portugal, Oct. 2009. - P. Lopes, J. Arrais and J. L. Oliveira
“Dynamic Service Integration using Web-based Workflows”
In 10th International Conference on Information Integration and Web Applications & Services“, Linz, Austria, Nov. 2008, pp. 622-625. - D. Santos, C. Costa, J. L. Oliveira, and A. Neves
“Alternative lossless compression algorithms in X-ray cardiac images” in Computational Vision and Medical Image Processing, N. J. João Tavares, Ed. London: Taylor & Francis Group, 2008, pp. 143-146. - D. Polónia, C. Costa, and J. L. Oliveira
“A Model to Optimize the Use of Imaging Equipment and Human Skills Scattered in Very Large Geographical Areas”
In 9th International Conference on Enterprise Information Systems (ICEIS 2004), Madeira, Portugal, 2007. - V. Afreixo, A. Freitas, M. Pinheiro, J. L. Oliveira, G. Moura, and M. A. S. Santos
“Exploiting a Biclustering algorithm in ORFeome analysis”
In Proceedings of the 2007 VLDB Workshop on Data Mining in Bioinformatics, Vienna, Austria, 2007. - A. Freitas, J. Duarte, M. Pinheiro, J. L. Oliveira, G. Moura, and M. A. S. Santos
“Homo sapiens versus Pan troglodytes: quão diferentes são?” in Actas do XIV Congresso Nacional da Sociedade Portuguesa de Estatística, Covilhã, Portugal, 2007. - J. Duarte, A. Freitas, M. Pinheiro, J. L. Oliveira, G. Moura, and M. A. S. Santos
“ISA: um algoritmo de bi-classificação?” in Actas do XIV Congresso Nacional da Sociedade Portuguesa de Estatística, Covilhã, Portugal, 2007. - A. Freitas, M. Pinheiro, V. Afreixo, J. Duarte, J. L. Oliveira, G. Moura, and M. A. Santos
“A median-based Iterative Signature Algorithm”
In IASC 07 – Statistics for Data Mining, Learning and Knowledge Extraction, Aveiro, Portugal, 2007. - A. Freitas, M. Pinheiro, J. L. Oliveira, G. Moura, and M. A. Santos
“A new limiting distribution for a statistical test for the homogeneity of two multinomial populations”
In Workshop in Statistic on Genomics and Proteomic, CIM, 2006. - J. Arrais, D. Polónia, and J. L. Oliveira
“A prospective study on the integration of microarrays data in HIS/ERP”
In Biological and Medical Data Analysis (ISBMDA’ 2006), Lecture Notes in Computer Science, Vol. 4345, Thessaloniki, Greece, 2006. - D. Polónia, C. Costa, and J. L. Oliveira
“Optimizing PACS and imaging resources”
In The XX International Congress of the European Federation for Medical Informatics (MIE 2006), Maastricht, Netherlands, 2006. - G. Dias, J. L. Oliveira, F. Vicente, and F. Martín-Sanchez
“Integrating Medical and Genomic Data: a Sucessful Example for Rare Diseases”
In The XX International Congress of the European Federation for Medical Informatics (MIE 2006), Maastricht, Netherlands, 2006. - J. Arrais, J. L. Oliveira, G. Grimes, S. Moodie, K. Robertson, and P. Ghazal
“Microarray data sharing in BioMedicine”
In The XX International Congress of the European Federation for Medical Informatics (MIE 2006), Maastricht, Netherlands, 2006. - D. Polónia, C. Costa, and J. L. Oliveira
“A PACS based GRID of resources”
In 4th International EuroPACS Conference (EuroPACS 2006), Trondheim, Norway, 2006. - G. Dias, J. L. Oliveira, F. Vicente, and F. Martin-Sanchez
“Integration of Genetic and Medical Information Through a Web Crawler System”
In Biological and Medical Data Analysis (ISBMDA’ 2005), Lecture Notes in Computer Science, Vol. 3745, Aveiro, Portugal, 2005. - C. Costa, J. L. Oliveira, A. Silva, V. Ribeiro, and J. Ribeiro
“Data Management and Visualization Issues in a Fully Digital Echocardiography Laboratory”
In Biological and Medical Data Analysis (ISBMDA’ 2005), Lecture Notes in Computer Science, Vol. 3745, Aveiro, Portugal, 2005. - C. Costa, J. L. Oliveira, A. Silva, V. Ribeiro, and J. Ribeiro
“A Telemedicine Platform for Cardiovascular Ultrasound”
In The XIX International Congress of the European Federation for Medical Informatics (MIE 2005), Geneve, Switzerland, 2005. - D. Polónia, C. Costa, and J. L. Oliveira
“Architecture evaluation for the implementation of a Regional Integrated Electronic Health Record”
In The XIX International Congress of the European Federation for Medical Informatics (MIE 2005), Geneve, Switzerland, 2005. - M. Pinheiro, J. L. Oliveira, M. A. S. Santos, H. Rocha, M. L. Cardoso, and L. Vilarinho
“Results of a Biomedical Application in Newborn Screening Programs”
In The 3rd European Medical and Biological Engineering Conference (EMBEC 2005), Prague, Czech Republic, 2005. - J. Arrais, L. Silva, M. Rodrigues, L. Carreto, J. L. Oliveira, and M. A. S. Santos
“Why Another Microarrays LIMS”
In The 3rd European Medical and Biological Engineering Conference (EMBEC 2005), Prague, Czech Republic, 2005. -
D. Polónia, J. L. Oliveira, and N. P. Rocha
“Overview of information systems training in Portuguese medicine courses”
In Health and Medical Informatics Applications – Educational Aspects (EFMI-STC 2005), Athens, Greece, 2005. -
D. Polónia and J. L. Oliveira
“Health Information Systems and Telematics: A best of breed evaluation framework for the Portuguese case”
In European Health Management Association Annual Conference 2005 (EHMA 2005), Barcelona, Spain, 2005. -
F. Vicente, I. Hermosilla, M. García-Remesal, D. Pérez del Rey, B. Romero, I. Oliveira, J. L. Oliveira, A. S. Pereira, and F. Martin-Sanchez
“Infogenmed: Un Laboratorio Virtual para la Integración de Información Clínica y Genética en Aplicaciones Médicas”
In VII Congreso Nacional de Informático de la Salud (Inforsalud 2004), Madrid, Spain, 2004. -
Oliveira, J. L. Oliveira, F. Martin-Sanchez, V. Maojo, and A. S. Pereira
“Biomedical information integration for health applications with Grid: a requirements perspective”
In Healthgrid 2004, Clermont-Ferrand, France, 2004. -
D. Polónia, J. L. Oliveira, and M. O. Duarte
“Information Systems (IS) in the Third and Fourth Generation Mobile Operator”
In 5ª Conferência da Associação Portuguesa de Sistemas de Informação (CAPSI 2004), Lisbon, 2004. -
C. Costa, A. Silva, J. L. Oliveira, V. Ribeiro, and J. Ribeiro
“Himage PACS: A new approach to storage, integration and distribution of cardiologic images”
In Progress in Biomedical Optics and Imaging, vol. 5, H. H. Ratib OM, Ed., SPIE, 2004, pp. 277-287. -
M. Pinheiro, J. L. Oliveira, M. A. S. Santos, H. Rocha, M. L. Cardoso, and L. Vilarinho
“NeoScreen: A software application for MS/MS newborn screening analysis”
In International Symposium on Biological and Medical Data Analysis (ISBMDA 2004), Lecture Notes in Computer Science, Vol. 3337, Barcelona, Spain, 2004. -
J. L. Oliveira, G. Dias, I. Oliveira, P. Rocha, I. Hermosilla, F. Vicente, I. Spiteri, F. Martin-Sanchez, and A. S. Pereira
“DiseaseCard: a web-based tool for the collaborative integration of genetic and medical information”
In Biological and Medical Data Analysis (ISBMDA 2004), Lecture Notes in Computer Science, Vol. 3337, Barcelona, Spain, 2004. -
D. Polónia, I. Oliveira, and J. L. Oliveira
“A Business Process Model for Public Health Information Systems: A Governmental Perspective”
In 6th International Conference on Enterprise Information Systems (ICEIS 2004), Porto, Portugal, 2004. -
C. Costa, J. L. Oliveira, and A. Silva
“E-Services in Mission-Critical Organizations: Identification Enforcement”
In 6th International Conference on Enterprise Information Systems (ICEIS 2004), Porto, Portugal, 2004. -
M. García-Remesal, V. Maojo, H. Billhardt, J. Crespo, R. Alonso-Calvo, D. Pérez, F. Martín, V. López, J. Sánchez, F. Vicente, M. García-Rojo, A. Gómez de la Cámara, A. Sousa, J. L. Oliveira, I. Oliveira, M. Santos, and A. Babic
“Designing New Methodologies for Integrating Biomedical Information in Clinical Trials”
In EuroMISE 2004, Prague, Czech Republic, 2004. -
C. Costa, A. Silva, J. L. Oliveira, A. S. Pereira, and V. Ribeiro
“A New Concept for an Integrated Healthcare Access Model”
In Medical Informatics Europe (MIE 2003), Saint Malo, France, 2003. -
C. Costa, J. L. Oliveira, and A. Silva
“Authentication Model to Enforce Network Entities Identification”
In 4rd Conference on Telecommunications (ConfTele 2003), Aveiro, Portugal, 2003. -
C. Costa, J. L. Oliveira, A. Silva, and V. Gama
“An Integrated Access Interface to Multimedia EPR”
In 21st International EuroPACS Meeting (EuroPACS 2003), London, United Kingdom, 2003. -
Oliveira, J. L. Oliveira, M. Santos, F. Martin-Sanchez, and A. S. Pereira
“On the requirements of biomedical information tools for health applications: the INFOGENMED case study”
In 7th Portuguese Conference on Biomedical Engineering (BioEng 2003), Lisbon, Portugal, 2003. -
C. Costa, J. L. Oliveira, and A. Silva
“Electronic Patient Record Virtually Unique based on a Crypto Smart Card”
In International Conference on Web Engineering (ICWE 2003), Lecture Notes in Computer Science, Vol. 2722, Oviedo, Spain, 2003. -
C. Costa, J. L. Oliveira, and A. Silva
“Critical Information Systems Authentication based on PKC and Biometrics”
In International Conference on Web Engineering (ICWE 2003), Lecture Notes in Computer Science, Vol. 2722, Oviedo, Spain, 2003. -
C. Costa, J. L. Oliveira, and A. Silva
“A User-Oriented Model to Manage Multiple Digital Credentials”
In IEEE 5th International Conference on Enterprise Information Systems (ICEIS 2003), Angers, France, 2003. -
J. C. Santos, J. L. Oliveira, and C. Costa
“A User-oriented Multi-service Access Control System”
In 5ª Conferência sobre Redes de Computadores (CRC 2002), Faro, Portugal, 2002. -
C. Costa, J. L. Oliveira, and A. Silva
“A Trusted Brokering Service for PKI Interoperability and Thin-Clients Integration”
In IEEE Third International Conference on Enterprise Information Systems (ICEIS 2001), Setúbal, Portugal, 2001. -
C. Costa, J. L. Oliveira, and A. Silva
“Um Novo Mecanismo de Autenticação para Sistemas de Informação Clínica” (in portuguese)
In 4ª Conferência sobre Redes de Computadores (CRC 2001), Covilhã, Portugal, 2001.
Abstracts
- P. M. Coloma, M. J. Schuemie, G. Trifirò, L. Furlong, E. V. Mulligen, A. Bauer-Mehren, P. Avillach, J. Kors, F. Sanz, J. Mestres, J. L. Oliveira, S. Boyer, E. A. Helgee, M. Molokhia, J. Matthews, D. Prieto-Merino, R. Gini, R. M. C. Herings, G. Mazzaglia, G. Picelli, L. Scotti, L. Pedersen, J. V. d. Lei, and M. C. J. M. Sturkenboom, “Triage and Evaluation of Potential Safety Signals Identified from Electronic Healthcare Record Databases,” in 12th Annual Meeting of the International Society of Pharmacovigilance (ISoP 2012), Cancun, Mexico, 2012.
- P. Lopes and J. L. Oliveira, “COEUS: “Semantic Web in a box” for biomedical applications,” in 11th European Conference on Computational Biology (ECCB 2012), Basel, Switzerland, 2012.
- P. Lopes, D. Campos, T. Nunes, and J. L. Oliveira, “Delivering advanced pharmacovigilance with the EU-ADR Web Platform,” in 11th European Conference on Computational Biology (ECCB 2012), Basel, Switzerland, 2012.
- M. J. Correira, N. Rosa, I. Silveira, J. Arrais, J. L. Oliveira, and M. Barros, “Oral microbial proteome: clues of OralCard”, in 22nd IUBMB & 37th FEBS Congress, Seville, Spain, 2012.
- N. Rosa, J. Arrais, J. Melo, E. Coelho, M. J. Correira, J. L. Oliveira, and M. Barros, “OralCard: a bioinformatic tool dedicated to the oral cavity system”, in 22nd IUBMB & 37th FEBS Congress, Seville, Spain, 2012.
- E. Molero, C. Diaz, F. Sanz, J. L. Oliveira, G. Trifirò, A. Fourrier, M. Molokhia, L. Pedersen, S. Boyer, L. Scotti, R. Gini, R. Herings, C. Giaquinto, M. I. Loza, G. Mazzaglia, J. v. d. Lei, and M. Sturkenboom, “The EU-ADR Alliance: A Federated Collabora- tive Framework for Drug Safety Studies,” in 12th Annual Meeting of the International Society of Pharmacovigilance (ISoP 2012), Barcelona, Spain, 2012.
- P. Lopes, J. Arrais, and J. L. Oliveira, “A Knowledge Interoperability Framework for Integrating Genotype-to-Phenotype Data”, in III Xornadas Galegas de Bioinformatics (XGB 2012), Vigo, Spain, 2011.
- D. Campos, S. Matos, and J. L. Oliveira, “A machine learning-based tool for biomedical entity recognition (Best Poster)”, in III Xornadas Galegas de Bioinformatics (XGB 2012), Vigo, Spain, 2011.
- J. Arrais and J. L. Oliveira, “Using biomedical networks to gene-disease indentification”, in III Xornadas Galegas de Bioinformatics (XGB 2012), Vigo, Spain, 2011.
- P. Lopes and J. L. Oliveira, “An Integrated View for Human Variome Information,” in 14th International Conference on Research in Computational Molecular Biology (RECOMB 2010), Lisbon, Portugal, 2010.
- P. Gaspar, J. L. Oliveira, J. Frommlet, G. Moura, and M. A. S. Santos, “A gene multi-optimization tool as novel approach to heterologous expression,” in 14th International Conference on Research in Computational Molecular Biology (RECOMB 2010), Lisbon, Portugal, 2010.
- J. Frommet, P. Gaspar, M. Pinheiro, J. L. Oliveira, A. C. Gomes, G. Moura, and M. A. S. Santos, “Integrating tRNA abundance, codon-usage, codon-context and functional rare codon data in de novo synthesis of Plasmodium falciparum genes for optimal expression in Escherichia coli,” in 23rd tRNA Workshop (tRNA 2010), Aveiro, Portugal, 2010.
- S. Matos, J. Arrais, J. Maia-Rodrigues, and J. L. Oliveira, “QuExT: a concept-based query expansion approach for literature retrieval from MEDLINE”, in Jornadas de Bioinformática (JB 2009), Lisbon, Portugal, 2009.
- P. Lopes and J. L. Oliveira, “Integration of Variome Data Through a Link Discovery Strategy”, in Jornadas de Bioinformática (JB 2009), Lisbon, Portugal, 2009.
- A. Freitas, V. Afreixo, M. Pinheiro, J. L. Oliveira, and M. A. S. Santos, “Uma avaliação da performance do algoritmo de bi-classificação ISA-mediana”, in XVI Jornadas de Classificação e Análise de Dados (JOCLAD 2009), Faro, Portugal, 2009.
- S. Matos, A. Barreiro, “Syntactic-semantic analysis for information extraction in biomedicine”, NooJ Conference 2009, Jozeur, Tunisia, Jun 2009.
- Maria dos Santos, Miguel Pinheiro, Manuel A. S. Santos (2008). Candida albicans as a model system to study genetic code alterations at genome level. 9th ASM Conference on Candida and Candidiasis, Jersey City, USA (Mar 24-28)
- M. Santos, M. Pinheiro, J. L. Oliveira and M. A. S. Santos, “Comparative tRNomics of a genetic code alteration”, ESF-EMBO Symposium on Comparative Genomics of Eukaryotic Microorganisms: Eukaryotic Genome Evolution, Sant Feliu de Guixols, Spain, 2007.
- M. Pinheiro, G. Moura, A. Freitas, M. A. S. Santos, and J. L. Oliveira, “A bioinformatics system to analyze sequences at a genomic scale”, in XVth National Congress of Biochemistry (NBC 2006) Aveiro, Portugal, 2006.
- G. Moura, M. Pinheiro, C. Gomes, A. Freitas, J. L. Oliveira, and M. A. S. Santos, “Large Scale Comparative Codon Context Analysis Unveils Novel Rules Governing Gene Evolution”, in XVth National Congress of Biochemistry (NBC 2006) Aveiro, Portugal, 2006.
- J. P. Lousado, G. Moura, M. Pinheiro, J. L. Oliveira, and M. A. S. Santos, “Large Scale Comparative Genomics of Codon Context”, in XVth National Congress of Biochemistry (NBC 2006) Aveiro, Portugal, 2006.
- S. Lima, E. Fonseca, J. L. Oliveira, and C. Egas, “Computational Methodologies for Metagenomics Projects”, in XVth National Congress of Biochemistry (NBC 2006) Aveiro, Portugal, 2006.
- C. Gomes, G. Moura, A. C. Gomes, J. L. Oliveira, M. Pinheiro, A. Freitas, and M. A. S. Santos, “The role of codon context on mRNA decoding error in vivo in yeast”, in XVth National Congress of Biochemistry (NBC 2006) Aveiro, Portugal, 2006.
- A. Freitas, L. Ferreira, J. Duarte, M. Pinheiro, J. L. Oliveira, G. Moura, and M. A. S. Santos, “Homo Sapiens Vs Pan Troglodytes: How Diferent Are They?” in XVth National Congress of Biochemistry (NBC 2006) Aveiro, Portugal, 2006.
- C. Egas, J. L. Oliveira, M. A. S. Santos, M. S. Costa, and C. Faro, “Whole Genome shotgun mapping of Rubrobacter radiotolerans”, in XVth National Congress of Biochemistry (NBC 2006) (Invited talk), Aveiro, Portugal, 2006.
- L. Carreto, P. Pereira, J. Arrais, J. L. Oliveira, and M. A. S. Santos, “National Facility For DNA Microarrays”, in XVth National Congress of Biochemistry (NBC 2006) (Invited talk), Aveiro, Portugal, 2006.
- J. Arrais, J. L. Oliveira, G. Campos, L. Carreto, and M. A. S. Santos, “Microarray data: from the hybridisation to the analysis”, in European Summer School in Biomedical Informatics, Balatonfüred, Hungary, 2006.
- J. Arrais, L. Carreto, M. A. S. Santos, and J. L. Oliveira, “Collaborative work on microarrays using MAGE-ML”, in 9th International Meeting of the Microarray Gene Expression Data Society (MGED9), Seattle, Washington, USA, 2006.
- J. Arrais, L. Carreto, H. Pais, F. Lopes, M. A. S. Santos, and J. L. Oliveira, “A Laboratory Information Management System for Microarray Data Storage, Sharing and Analysis”, in XVth National Congress of Biochemistry (NBC 2006) Aveiro, Portugal, 2006.
- P. R. Almeida, L. Carreto, J. L. Oliveira, and M. A. S. Santos, “Software-Assisted Design of Probes for SNPs Detection using Oligonucleotide Microarrays: Applications in a Case Study”, in XVth National Congress of Biochemistry (NBC 2006) Aveiro, Portugal, 2006.
- J. Arrais, J. L. Oliveira, G. Campos, L. Carreto, and M. A. S. Santos, “Microarray data: from the hybridisation to the analysis”, in European Summer School in Biomedical Informatics, Balatonfüred, Hungary, 2006.
- J. Arrais, L. Carreto, M. A. S. Santos, and J. L. Oliveira, “Collaborative work on microarrays using MAGE-ML”, in 9th International Meeting of the Microarray Gene Expression Data Society (MGED9), Seattle, Washington, USA, 2006.
- L. Carreto, M. C. Santos, J. Arrais, M. Rodrigues, L. Silva, J. L. Oliveira, and M. A. S. Santos, “Implementation of a National Facility for DNA Microarrays in Portugal”, in 8th International Meeting of the Microarray Gene Expression Data Society (MGED8), Bergen, Norway, 2005.
- J. L. Oliveira, M. Pinheiro, M. A. S. Santos, H. Rocha, M. L. Cardoso, and L. Vilarinho, “On the Processing of MS/MS Data: an Application for Metabolic Diseases Screening”, in XIV Congresso Nacional de Bioquímica, Vilamoura, Portugal, 2004.
- M. Pinheiro, V. Afreixo, G. Moura, A. Freitas, M. A. S. Santos, and J. L. Oliveira, “Bioinformation System for Unveil General Rules of Codon Context”, in XIV Congresso Nacional de Bioquímica, Vilamoura, Portugal, 2004.
- G. Moura, M. Pinheiro, V. Afreixo, A. Freitas, J. L. Oliveira, and M. A. S. Santos, “Comparative codon context analysis in complete genomes unveils new decoding rules in yeast”, in XIV Congresso Nacional de Bioquímica, Vilamoura, Portugal, 2004.
- G. Moura, M. Pinheiro, V. Afreixo, A. Valente, J. L. Oliveira, and M. A. S. Santos, “Comparative codon context analysis at a genomic scale defines new mRNA decoding rules”, in Translational Control Meeting, Cold Spring Harbor, New York, 2004.
- L. Carreto, J. Pereira, J. L. Oliveira, A. S. Pereira, V. Afreixo, A. Valente, and M. A. S. Santos, “Implementation of a DNA-microarray platform for yeast expression, genome diversity and evolution and molecular diagnostics research.” in XII Jornadas de Biologia de Leveduras (Leveduras’2004), Aveiro, Portugal, 2004.
- G. Moura, M. Pinheiro, V. Afreixo, A. Valente, J. L. Oliveira, and M. A. S. Santos, “Genome scale codon context analysis for Saccharomyces cerevisiae and Candida albicans.” in XII Jornadas de Biologia de Leveduras (Leveduras’2004), Aveiro, Portugal, 2004.
- M. Pinheiro, V. Afreixo, G. Moura, G. Dias, A. S. Pereira, A. Valente, M. A. S. Santos, and J. L. Oliveira, “Bioinformation system and statistical methodologies for gene primary structure analysis”, in EuroMISE 2004, Prague, Czech Republic, 2004.
- J. L. Oliveira, M. Pinheiro, G. Dias, G. Moura, V. Afreixo, A. Valente, A. S. Pereira, and M. A. S. Santos, “New software tools for gene analysis”, in ESF Programme in Functional Genomics: 1st European Conference (ESFFG’2003), Prague, Czech Republic, 2003.
- Oliveira, A. Furtado, J. L. Oliveira, A. S. Pereira, V. Maojo, F. Martin-Sanchez, A. Babic, and M. Santos, “Integration of Genetic and Clinical Information Sources for Health Applications”, in ESF Programme in Functional Genomics: 1st European Conference (ESFFG’2003), Prague, Czech Republic, 2003.
- J. L. Oliveira, G. Dias, R. Silva, and M. A. S. Santos, “An open environment for management of proteomics data and projects”, in ESF Programme in Functional Genomics: 1st European Conference (ESFFG’2003), Prague, Czech Republic, 2003.
Thesis
- Eduardo Pinho
Title: Multimodal information retrieval in medical imaging repositories
Supervisor: Carlos Costa
Date: 2019 - Fernanda Brito Correia
Title: Prediction And Analysis Of Biological Networks Structure And Dynamics
Supervisor: José Luís Oliveira, Joel Arrais
Date: 2019 - Tiago Godinho
Title: Performance Optimization in Medical Imaging Networks
Supervisor: Carlos Costa
Date: 2018 - Pedro Sernadela
Title: Semantic Knowledge Discovery for Neuropathologies
Supervisor: José Luís Oliveira
Date: 2018 - Edgar Coelho
Title: Computational prediction of Inter-Species Protein-Protein Interactions
Supervisor: José Luís Oliveira, Joel Arrais
Date: 2017 - Eriksson Monteiro
Title: Distributed and Interactive Solutions for Ubiquitous Telemedicine
Supervisor: José Luís Oliveira, Carlos Costa
Date: 2017 - Luís Bastião Silva
Title: A federated architecture for biomedical data integration
Supervisor: Carlos Costa, José Luís Oliveira
Date: 2015 - Paulo Gaspar
Title: Computational methods for gene characterization and genome knowledge extraction
Supervisor: José Luis Oliveira, Sérgio Matos
Date: 2015 - Luis Ribeiro
Title: Distributed Medical Imaging Repositories
Supervisor: Carlos Costa
Date: 2014 - David Campos
Title: Term expansion methodologies in biomedical information retrieval
Supervisor: José Luis Oliveira, Sérgio Matos
Date: 2014 - Tiago Pedrosa
Title: Electronic Health Records for Mobile Citizens
Supervisor: José Luis Oliveira, Rui Pedro Lopes
Date: 2013 - Pedro Lopes
Title: Service Composition in Biomedical Applications
Supervisor: José Luis Oliveira
Date: 2012 - Nuno Rosa
Title: From the oral cavity proteome to the proteome
Supervisor: Marlene Barros, José Luis Oliveira
Date: 2012 - Daniel Ferreira Polónia
Title: An electronic market for teleradiology services
Supervisor: José Luis Oliveira, Manuel Oliveira Duarte
Date: 2011 - José Paulo Lousado
Title: Analysis of tandem repeats in DNA primary structures
Supervisor: José Luis Oliveira
Date: 2011 - Miguel Monsanto Pinheiro
Title: Computational systems for the study of the primary structure and redesign of genes
Supervisor: José Luis Oliveira, Manuel Santos
Date: 2010 - Joel Perdiz Arrais
Title: Microarrays information systems and healthcare information systems
Supervisor: José Luis Oliveira
Date: 2010 - Carlos Manuel Azevedo Costa
Title: A security dynamic model for healthcare information systems
Supervisor: José Luis Oliveira, Augusto Silva
Date: 2004
Developing new Bioinformatics tools for genome analysis
Funding entity: POCTI-32030/2001
Period: 2002-2005
Biologists have been wondering for many years how organisms evolved highly accurate information maintenance, transfer and decoding machineries. In particular, how the astonishing translational decoding rate of 20 codons per second is achieved with an average error of 10-4 to 10-5 per codon decoded, and how does the ribosome maintain the reading frame. The tools to answer these questions are not yet available but the row DNA sequencing data is. To shed new light into this important question, we have developed a software package that simulates ribosome scanning and reading during mRNA translation. The software screens fully or partially sequenced genomes and determines the arrangement of any particular codon in relation to the others by simultaneously fixing P-site codons and “memorizing” E and A-site codons during each translocation cycle. In doing so, it builds a genome wide codon context map that allows for identification of potential error prone mRNA sequences and gene expression regulatory points.
In this project, the various tools already developed will be integrated into a single software package to allow for automated search, downloading and editing of row DNA sequence data. Software tools for data display and new mathematical methodologies for identification of general rules governing mRNA translation will be developed. New tools for mapping mRNA regions of high decoding error and putative gene expression regulatory sequences present in the mRNAs, will also be developed. Finally, a database and an Internet Home Page will be built for making the data available to the scientific community. These in silico studies will be complemented with in vivo experiments. For this, a multidisciplinary team including two computing engineers, two mathematicians, one physicist, one biochemist and one molecular biologist has been assembled. To our knowledge this is the first Portuguese multidisciplinary team set up for functional genomics and the only one actively engaged on the development of software tools and mathematical models for genome analysis. It is expected that this project will provide important new insight on the role of the translational machinery on genome evolution.
Functional Proteomics in Candida albicans: Developing an Integrated Database for the Management of Proteomics projects
Funding entity: POCTI-32942/99
Period: 2001-2004
Candida albicans is an important human pathogen which exists as a commensal in at least 50% of the human population. It accounts for more than 60% of all fungal infections and is now the fourth most common form of septicaemia in Western hospitals with an associated mobidity between 30 and 50%. It is also a major cause for concern in HIV-infected populations where 84% of the patients develop oropharyngeal C.albicans colonisation and 55% develop clinical thrush. C. albicans pathogenesis is dependent upon a wide range of virulence factors, namely a myriad of morphogenesis associated factors, represents a major challenge to the elucidation of C. albicans pathogenesis at the molecular level through classic molecular and biochemical methodologies. The diploid nature of C. albicans, its alternative genetic code and its recalcitrance to genetic analysis, add extra difficulties to its study and to the development of new antifungals. However, the advent of new genetics and molecular technologies which allow for genome wide analysis is promising to alter the present situation.
This project aims at integrating classical genetics and biochemical approaches with newly developed, proteomics and bioinformatics methodologies to uncover new virulence factors associated to morphogenesis.
Software tools are been developed for management of biological data extracted from protein 2D-maps, for helping planning and following up experimental protocols and for data storing. Additionally, mathematical algorithms are also been developed for creating theoretical protein 2D-maps for comparative proteomics studies.
Query term expansion methodologies for improved biomedical literature retrieval
Funding entity: FCT PTDC/EIA-CCO/100541/2008
Period: 2010-2013
The objective of this project is to develop a query expansion and document ranking method specially aimed at obtaining, from the MEDLINE database, a ranked list of publications that are most significant to a set of genes.
Faculty
- Alina Trifan
- Armando Pinho
- Carlos Costa
- Diogo Pratas
- Joel Arrais
- José Luís Oliveira
- Olga Fajarda
- Raquel Silva
- Sérgio Matos
Post-docs
- Sérgio Matos
- Joel Arrais
PhD Students
- Arnaldo Pereira
- João Figueira Silva
- João Rafael Almeida
- Jorge Miguel Silva
- Micael Pedrosa
- Nelson Monteiro
- Rodrigo Escobar
- Rui Antunes
- Rui Lebre
- Sara Duarte Pereira
- Tiago Almeida
- Yubraj Gupta
MSc Students
- André Pedrosa
- Lucas Silva
- Lucas Barros
- João Ribeiro
- Pedro Cavadas
- Pedro Oliveira
- Pedro Matos
- Mariana Sequeira
Hello world!
Welcome to WordPress. This is your first post. Edit or delete it, then start blogging!
DICOM Services Over Peer-To-Peer Networks
Funding entity: FCT PTDC/EIA-EIA/104428/2008
Period: 2010-2013
The overall goal is to instantiate a new network connectivity concept for medical imaging data and services at inter-institutional level. This will turn large volumes of clinical information and analytical tools, actually “locked” in clinical units, into shared repositories and high-quality collaborative environments for medical applications, education and research.
EU-ADR project in ua_online
IEETA explora potencialidades das TIC na detecção precoce de reacções adversas a medicamentos
ua_online
GEN2PHEN – Genotype-To-Phenotype Databases: A Holistic Solution
Funding entity: FP7-Health (IP)
Period: 2008-2012
The GEN2PHEN project has the overall ambition of unifying human and model organism genetic variation databases, and doing this in such a way that the resulting holistic view of G2P data can be blended with all other biomedical database domains via one or more central genome browsers.

OM
Organization Measurement (OM) method
About
The complete protein-protein interaction (PPI) network of even the most studied organisms is yet to be fully established. This is mostly due to lack of reliability and accuracy of the high-throughput experimental methods used for PPI identification. PPIs can be conveniently represented as networks, allowing the use of graph theory in their study. Different network-based methods have been used to identify false-positive interactions and missing links in biological networks. Network topology studies may reveal patterns associated with specific organisms or the type of PPIs. Thus, in this paper, we propose a new methodology to denoise PPI networks and predict missing links solely based on the network topology, the Organization Measurement (OM) method.
The OM methodology was applied in the denoising of Saccharomyces cerevisiae (Yeast and CS2007) and Homo sapiens (Human). To evaluate our methodology, two strategies were used. The first compared its application in random networks and in the gold standard networks, while the second perturbed the networks with the gradual random addition and removal of edges. The applied validation strategy showed that the proposed methodology achieves an AUC of 0.95 and 0.87, in Yeast and Human networks, respectively. The random removal of 80% of the Yeast gold standard interactions resulted in an AUC of 0.71, whereas the random addition of 80% interactions resulted in an AUC of 0.75. In Human, the random removal of 80% interactions resulted in an AUC of 0.62, while the random addition of 80% interactions resulted in an AUC of 0.72. Applying the OM methodology to the CS2007 dataset yields an AUC of 0.99. We also perturbed the network of the CS2007 dataset by randomly inserting and removing edges in the same proportions previously described. The percentage of false positives identified and removed from the network varied from 97%, when inserting 20% more edges, to 89% when 80% more edges were inserted. The percentage of true positives identified and inserted in the network varied from 95% when removing 20% of the edges, to 40% after the random deletion 80% edges.
The implemented tests show that the OM methodology is sensitive to the topological structure of the biological networks and can be used for network denoising. The obtained results suggest that the present approach can efficiently denoise PPI networks and that it can be applied to different organisms, as long as they have inherent patterns in the structures of their network models. In addition, although the performance of the method correlates with the initial quality of the network, improvements were consistently obtained.
QuARC
HighFCM
 Exploring deep Markov models in genomic data compression using sequence pre-analysis
Exploring deep Markov models in genomic data compression using sequence pre-analysis
AboutHighFCM is a compression algorithm that relies on a pre-analysis of the data before compression, with the aim of identifying regions of low complexity. This strategy enables to use deeper context models, supported by hash-tables, without requiring huge amounts of memory. As an example, context depths as large as 32 are attainable for alphabets of four symbols, as is the case of genomic sequences. These deeper context models show very high compression capabilities in very repetitive genomic sequences, yielding improvements over previous algorithms. Furthermore, this method is universal, in the sense that it can be used in any type of textual data (such as quality-scores). HighFCM was designed and implemented at IEETA, a research unit of the University of Aveiro, and is available for non-commercial use.
CitationDiogo Pratas and Armando J. Pinho. “Exploring deep Markov models in genomic data compression using sequence pre-analysis”. Proc. of the European Signal Processing Conference, EUSIPCO 2014, Lisboa, Portugal, September 2014.
DOI: to add.
Downloadoralint
Interactome for the Human oral cavity
About
From birth, humans are subject to the colonization and invasion attempts of numerous microorganisms. Although in normal situations, contacting with microbes can support the shaping and development of our immune system, specific situations, such as stress or an unhealthy diet, can render us vulnerable to opportunistic pathogens.
Since the oral cavity is particularly exposed to the environment, it is an anatomic region prone to microbial invasion. Additionally, one of the requirements for bacterial colonization and cellular invasion is the establishment of protein-protein interactions (PPIs) with the host. With this in mind, we aim to develop a computational method for prediction of the oral human-microbial interactome.
Revealing the human-microbial interactome will allow further understanding of the mechanisms behind the onset of oral diseases. Additionally, this knowledge may give insight on key proteins involved in oral infections, which can be used for either diagnosis, as molecular biomarkers, or for treatment, as drug-targets.
Download data
Oral proteins from proteomic studies
Dataset train/validation
Interactome – obtained predictions
Cytoscape project
DTIpred
Computational Discovery of Putative Leads for Drug Repositioning Through Drug-Target Interaction Prediction
About
The emergence of multi-resistant bacterial strains and the existing void in the discovery and development of new classes of antibiotics is a growing concern, as some bacterial strains are now resistant to last-line antibiotics and considered untreatable. A growing trend in drug screening for the past decade is drug repositioning, which consists in focusing on one of the undesired effects of an already commercialized drug in an attempt to make it the main effect. While this was formerly performed experimentally, computational methods speed and reduce the associated costs of drug and drug-target screening.
Thus, we present a computational pipeline that enables the discovery of putative leads for drug repositioning that can be applied to any microbial proteome. Putative drug-targets are inferred by calculating network metrics for the interactome of the bacterial organism. Prediction of drug-target interactions (DTI) is performed using a random forest trained with high-quality publicly available data. Classifier performance achieved an area under the ROC curve of 0.91 for classification of out-of-sampling data. A drug-target network was created by combining 3,081 unique ligands and the expected ten best drug targets. This network was used to predict new DTIs and to calculate the probability of the positive class, allowing the scoring of the predicted instances.
Available data (download ZIP)
– classifier.py (classifier code: uses Yamanishi’s data for training and DrugBank data for external validation)
– yamanishi_DTIs_REAL_NEGS.txt (Training data set: Protein ID; Drug ID; True Label; + 702 features)
– drugbank_DTIs_REAL_NEGS.txt (External validation data set: Protein ID; Drug ID; True Label; + 702 features)
– test_data_sc_and_bc.txt (Test data set: Protein ID; Drug ID; True Label; + 702 features)
Mesh PPI classifier
AboutThe worldwide surge of multiresistant microbial strains has propelled the search for alternative treatment options. A key aspect to this task is to understanding the mechanisms by which specific pathogens colonize, survive and replicate within the host, which can be achieved through the study of protein-protein interactions. Despite the advances of laboratorial techniques, protein sequence-based computational models allow the screening of protein interactions between entire proteomes in a fast and inexpensive manner. These models are specially valuable due to the recent advances in sequencing metagenomic organisms, where only the protein sequence is available.
Here, we present an improved supervised machine learning model for the prediction of protein interactions based on the protein structure. We propose the usage of the discrete cosine transform as an efficient methodology of representing protein sequences and use categories extracted from physicochemical properties of amino acids.
For the classification task we use a mesh of hyper-specialised classifiers dedicated to the most relevant pairs of Gene Ontology molecular function annotations.
Based on an exhaustive evaluation that includes datasets with different configurations, cross-validation and out-of-sampling validation, the obtained results outscore the state-of-the-art for sequence-based methods. For the final mesh model using SVM with RBF, a consistent average AUC of 0.84 was attained.
Available data-datasets: datasets used for testing
-d1: 6702 protein interactions dataset (50% negative, 50% positive)
-dataset1_RNA.fasta: mRNA sequences for the proteins of the dataset
-negatome_3351.txt- negative interactions (examples of proteins that do not interact)
-positive_3351.txt- positive interactions
-d2: 10000 interactions dataset(50% negative, 50% positive)
-d2.fasta: mRNA sequences for the proteins of the dataset
-negatome_verified_random.txt – negative interactions (examples of proteins that do not interact)
-shuffled-yeast-positive-10k.txt – positive interactions
-d3: 20000 interactions dataset (50% negative, 50% positive)
-20k_negative_random:
-protein_sequences.fasta – negative interactions (examples of proteins that do not interact)
-shuffled-yeast-positive-10k.txt – positive interactions
-yeast_ppi_orig.txt – original protein-protein interactions from yeast
-dct_d2_rbf: script used for studying d2 with dct rbf method
-dct_random_d3: script used for DCT method with dataset 3
-dct_rbf_parameters: script used for studying rbf parameters for dct
-dct_rbf_parameters: script used for studying rbf execution time
-original_code: original script
-guo_d3: script used for studying guo with dataset 3
-shen_d2: script used for studying shen with dataset 2
-shen_d3: script used for studying shen with dataset 3
-shen time: script used for studying shen execution time
-guo time: script used for studying guo execution time
Download