José Luis Oliveira
Universidade de Aveiro, DETI / IEETA
3810-193 Aveiro, Portugal
jlo@ua.pt
(+351) 234 370 500
smash
 About
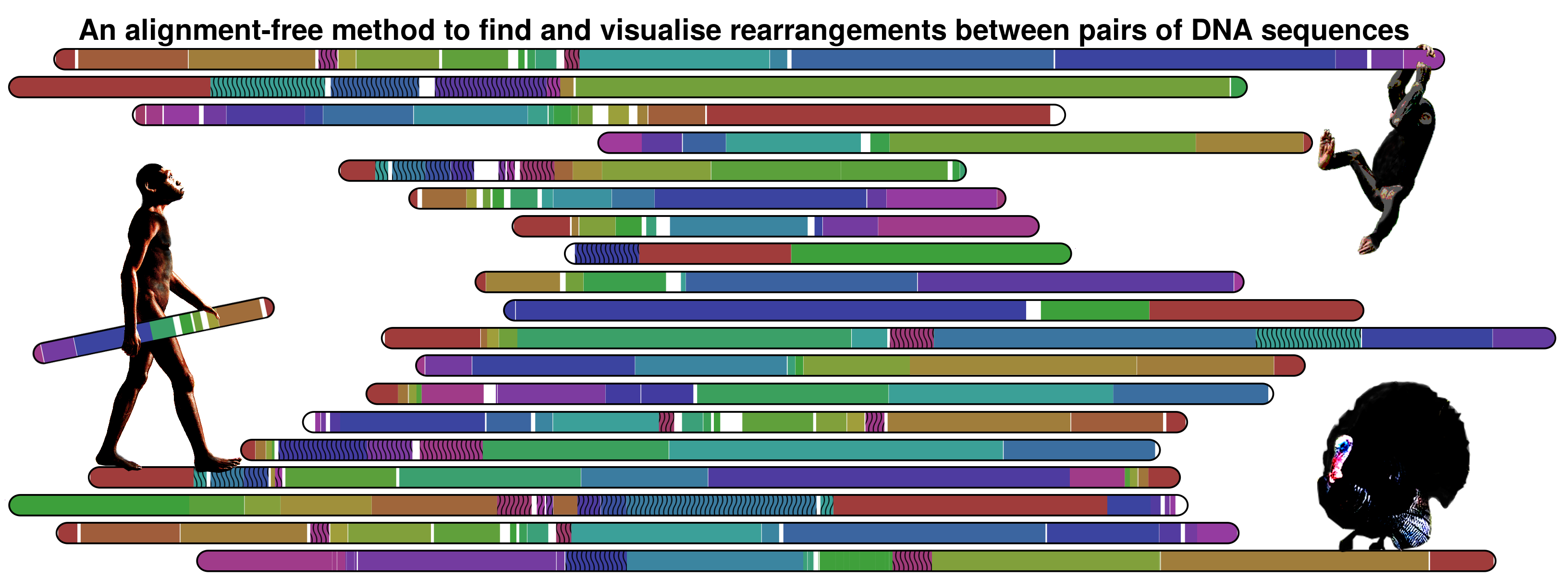
AboutSmash is a completely alignment-free method/tool to find and visualise genomic rearrangements. The detection is based on conditional exclusive compression, namely using a FCM (Markov model), of high context order (typically 20). For visualisation, Smash outputs a SVG image, with an ideogram output architecture, where the patterns are represented with several HSV values (only value varies). The method can perform both in small- and large-scale. Nevertheless is more directed to large-scale since that the main aim of the research is to know where the large-scale [chromosomal by chromosome] of several primates was equal/different, having at a glance a map of the entire genomes. Therefore the method aims to solve evolutionary species Rubik’s cube. The following image, illustrating the information maps between human and chimpanzee for the several chromosomes, depicts such an example study:


Nevertheless, the method is not limited to primates information. The following image show the information map between Meleagris gallopavo and Gallus gallus chromosomes 1 using a threshold of 0.95.
Download CitationDiogo Pratas, Raquel M. Silva, Armando J. Pinho, Paulo J. S. G. Ferreira. An alignment-free method to find and visualise rearrangements between pairs of DNA sequences. Sci. Rep. 5, 10203 (2015); doi:10.1038/srep10203.